
Abstract
Background:
No comparisons of different doses of docetaxel-capecitabine in patients with advanced gastric cancer have been performed.
Methods:
Patients with previously untreated metastatic/locally advanced gastro-oesophageal or gastric adenocarcinoma were enrolled in a prospective multicentre phase II trial. Two sequential cohorts received docetaxel 75 mg m−2 (day 1) plus capecitabine 1000 mg m−2 twice daily (days 1–14) (cohort I) or docetaxel 60 mg m−2 (day 1) plus capecitabine 800 mg m−2 twice daily (days 1–14) (cohort II) every 3 weeks. The primary end point was confirmed overall response rate.
Results:
In all, 91 patients were enrolled (cohort I, n=40; cohort II, n=51) and 87 were evaluable for efficacy (n=38, 49, respectively). Overall response rate was 50.0% in cohort I and 23.5% in cohort II (exploratory analysis, P=0.014). Median times to tumour progression and overall survival were 5.6 and 10.1 months in cohort I and 3.7 and 7.2 months in cohort II, respectively. Dose reductions for docetaxel and capecitabine were required in 50.0% and 57.5% of patients in cohort I and 11.8% and 15.7% in cohort II, respectively.
Conclusion:
Starting treatment with full doses and reducing promptly seems to be the more promisingly effective strategy than starting cautiously with lower doses. Docetaxel/capecitabine 75/2000 mg m−2 is a manageable, convenient outpatient combination with promising efficacy against advanced gastric cancer.
Similar content being viewed by others


Main
Recent epidemiological data indicate that 0.93 million new cases of gastric and gastro-oesophageal junction cancer are diagnosed annually worldwide (Kamangar et al, 2006). More than 60% of patients present with locally advanced or metastatic disease or develop metastases after initial resection (Parkin et al, 1999). In patients with advanced gastric cancer, chemotherapy improves overall survival compared with best supportive care (Wagner et al, 2010), but there is no single accepted standard chemotherapy regimen. Both cisplatin plus 5-fluorouracil (CF) (Kim et al, 1993; Vanhoefer et al, 2000; Ohtsu et al, 2003; van Cutsem et al, 2006) and epirubicin plus CF (ECF) (Webb et al, 1997; Ross et al, 2002) have established efficacy and are recognised reference regimens for comparative trials.
During the past decade, several new-generation cytotoxic agents, including capecitabine, irinotecan, oxaliplatin and docetaxel, have been investigated for the treatment of gastric cancer. The phase III V325 study (van Cutsem et al, 2006) established the efficacy of docetaxel as first-line therapy for advanced gastric cancer. The final results showed that adding docetaxel to the CF regimen (DCF) improved overall survival, response rate, time to disease progression and health-related quality of life, but was associated with a marked increase in haematological toxicity. As a result, various cooperative groups and institutions tested modified versions of the DCF regimen in phase II studies (Park et al, 2005; Thuss-Patience et al, 2005; Lorenzen et al, 2007; Al-Batran et al, 2008; Shah et al, 2010; Tebbutt et al, 2010). As part of this initiative, we demonstrated that a simplified regimen of docetaxel plus 5-fluorouracil had similar efficacy to ECF in patients with advanced gastric cancer in a randomised phase II study (Thuss-Patience et al, 2005), suggesting that cisplatin does not need to be a mandatory component of an effective combination.
As our study was conducted, two phase III trials have shown that the oral fluoropyrimidine, capecitabine, has at least equivalent efficacy to infused 5-fluorouracil when given as part of a combination regimen in patients with advanced gastric cancer (Cunningham et al, 2008; Kang et al, 2009). Several phase II trials have reported promising efficacy with the combination of docetaxel and capecitabine in patients with advanced gastric cancer (Park et al, 2004; Chun et al, 2005; Kim et al, 2005; Lorenzen et al, 2005; Giordano et al, 2006; Rosati et al, 2007; Lo et al, 2010; Tebbutt et al, 2010).
All approaches to optimise docetaxel–fluoropyrimidine-based regimens focus on modifying chemotherapy dose and application. As there are no phase IIl trials investigating this question, it is unclear whether or not changes in chemotherapy dose affect efficacy. To our knowledge, there has been no comparison of different doses of docetaxel–capecitabine. Therefore, in this study of the Arbeitsgemeinschaft Internistische Onkologie (AIO), we examined two sequential patient cohorts treated with different doses of docetaxel in combination with capecitabine to determine if maximal doses are really necessary in the palliative setting or if lower doses might have similar efficacy with better tolerability.
Materials and methods
Study design
This was a prospective, non-randomised, multicentre phase II trial. Patients were recruited sequentially to two separate cohorts and treated with different doses of docetaxel plus capecitabine; the first 40 patients received higher doses of docetaxel–capecitabine (cohort I) and the next 51 patients received lower doses (cohort II).
After completion of cohort I, it was decided to enrich the information gained by this trial by an amendment, lowering the dose of chemotherapy in a similar study population. Although the regimen tested in cohort I proved to be effective, safe and tolerable, frequent dose reductions were necessary and Common Toxicity Criteria (CTC) grade 1 and 2 toxicities were frequent, which were bothersome for the patient. As the treatment setting was palliative and benefit should be maximised by minimal side effects, an alternate strategy was investigated. Cohort II was initiated with a dose reduction of both drugs by 20% to investigate whether grade 1–4 side effects can be reduced, further dose adjustments can be avoided, but efficacy can be maintained. A reduction by 20% resulted in dose levels for each individual drug, which were investigated previously (Lee et al, 2008; Tebbutt et al, 2010).
The study was conducted according to the Declaration of Helsinki, registered in www.clinicaltrials.gov (no. NCT00142038) and the protocol was approved by the local ethics body. All patients provided written informed consent before study entry.
Patient population
Ambulatory patients aged 18–75 years with histologically confirmed, locally advanced or metastatic adenocarcinoma of the stomach or gastro-oesophageal junction were eligible. Measurable disease was required for study inclusion. No previous chemotherapy for advanced/metastatic disease or concurrent radiotherapy was allowed. Patients were required to have an Eastern Cooperative Oncology Group performance status of 2 or less (Karnofsky performance index of ⩾60%). Adequate bone marrow (leukocyte count >3.0 × 109 l−1, platelet count >100 × 109 l−1), renal (serum creatinine <1.25 × upper limit of normal and/or creatinine clearance >60 ml min−1 according to Cockroft–Gault formula) and liver (serum bilirubin <1.5 mg dl−1, alanine aminotransferase and aspartate aminotransferase ⩽3 × upper limit of normal) function was required. Patients with a co-existing secondary tumour (except basal cell carcinoma of the skin or carcinoma in situ of the cervix), uncontrolled infection, central nervous system metastases, recent major surgery (<2 weeks), anatomical or inflammatory intestinal problems, or an inability to take oral medication were excluded. The eligibility criteria for both study cohorts were identical.
Study evaluation
At baseline, clinical investigations were performed within 8 days, and tumour imaging within 21 days, before study inclusion. The baseline evaluation included a physical examination, assessment of medical history and symptoms, evaluation of performance status and routine blood analysis (haematology and biochemistry). During treatment, full blood count was repeated every week and all other parameters were re-evaluated before each chemotherapy cycle.
An electrocardiogram and abdominal computed tomography scan were performed in all patients at baseline, with further imaging if clinically indicated (i.e. bone scan, head computed tomography). During treatment, imaging of target lesions and non-target lesions was repeated every two cycles (every 6 weeks) using the same imaging procedures as at baseline. Tumour response was classified according to Response Evaluation Criteria In Solid Tumours 1.0 guidelines and verified 4–6 weeks later (Therasse et al, 2000), and evaluated by independent radiologists. After the end of study treatment, patients were followed up every second month until death. Toxicity was evaluated on a weekly basis using the National Cancer Institute-CTC (Version 2.0).
Tumour-related symptoms were assessed by the treating physician every 3 weeks on direct questioning. Symptomatic improvement was defined as subjective improvement as stated by the patient of at least one tumour-related symptom without worsening of any other or appearance of any new tumour-related symptom.
Treatment
Patients in cohort I received intravenous docetaxel 75 mg m−2 on day 1 given as a 1-h infusion plus oral capecitabine 1000 mg m−2 twice daily on days 1–14, every 3 weeks. Patients in cohort II received docetaxel 60 mg m−2 on day 1 and capecitabine 800 mg m−2 twice daily on days 1–14, every 3 weeks. Patients received chemotherapy for up to a maximum of 10 cycles, or until tumour progression or unacceptable toxicity.
Premedication with oral dexamethasone 8 mg was given the evening before day 1 of each cycle, as well as on the morning and evening of days 1 and 2. A 5-HT3 antagonist was also recommended on day 1. Prophylactic granulocyte-colony-stimulating factor was recommended in patients older than 70 years.
Treatment in both cohorts was delivered up to the beginning of toxicity and then interrupted or dose reduced: for grade 2 toxicities (except for isolated neutropenia/leukopenia/anaemia or alopecia), treatment was interrupted until symptoms had resolved to grade 0 or 1. For a second occurrence of grade 2 toxicity or a grade 3 event, treatment was interrupted until symptoms had resolved to grade 0 or 1 and docetaxel and capecitabine doses were reduced by 25%. For a second occurrence of grade 3 toxicity or a grade 4 event, treatment was interrupted until symptoms had resolved to grade 0 or 1 and docetaxel and capecitabine doses were reduced to 50%. Uncomplicated neutropenia/leukopenia grade 3 or 4 did not lead to dose reductions of docetaxel/capecitabine or interruption of capecitabine as long as it resolved to grade 1 before starting the next cycle. For grade 3/4 neutropenia/leukopenia lasting longer than 1 week or complicated by febrile neutropenia (body temperature ⩾38.5°C), the docetaxel dose was reduced by 25%. Granulocyte-colony-stimulating factor was not recommended as secondary prophylaxis in patients younger than 70 years.
Statistical analysis
The primary study end point was overall response rate, and secondary end points were overall survival, time to tumour progression and toxicity. Time to tumour progression was defined as the time from study inclusion to documented progressive disease. Overall survival was defined as the time from study inclusion to death from any cause. Symptomatic improvement was defined as subjective improvement of at least one tumour-related symptom (e.g., dysphagia, weight loss, loss of appetite, pain, abdominal fullness after meals) without worsening of any other symptom or appearance of any new tumour-related symptom. Kaplan–Meier methods were used to estimate time-to-event end points. Statistical comparisons of efficacy end points between the two patient cohorts were exploratory only. The statistical model was not prospectively designed for this comparison.
The study was designed according to the two-stage optimum design for phase II studies (Simon, 1989). For the null hypothesis, it was assumed that the true response rate was less than 25%, with an expected response rate of 45%. If at least 4 of the first 14 patients showed a response, the study was to be expanded to 44 patients. If at least 15 responses were documented in these 44 patients, the null hypothesis was rejected. This design provides a probability of 10% of accepting a regimen with a response rate of less than 25%, and a probability of 10% of rejecting a regimen with a response rate of more than 45%.
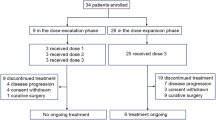
When the null hypothesis was rejected after 44 included patients (cohort I), the protocol was amended to continue with lower doses of docetaxel and capecitabine to optimise tolerability (cohort II). The same statistical design as for cohort I was applied for cohort II. The study was continued to a maximum of 95 patients (including drop outs) (see Figure 1).

Patient flowchart. *Reasons for exclusion: no gastric cancer (n=3) and patient gave wrong identity (n=1). **Because there were no drop outs, patient number exceeds that required statistically (n=44).
Results
In all, 95 patients were registered to the trial; four patients were excluded from the analysis owing to major violations of the inclusion criteria (see Figure 1). Therefore, a total of 91 evaluable patients were recruited from four centres in Germany from March 2004 to December 2006 (cohort I, n=40; cohort II, n=51). At the time of analysis (May 2008), 84 patients had died, six patients were alive and one patient was lost to follow-up.
Patient baseline characteristics are presented in Table 1. Baseline characteristics were generally well balanced between the two cohorts, except that patients in cohort I had more tumours of the gastro-oesophageal junction (50.0% vs 39.2% in cohort II) and more patients with >2 metastatic sites (57.5% vs 47.1%).
Treatment exposure
The total number of chemotherapy cycles administered in cohort I was 257 (median 6, range 1–10) and in cohort II was 247 (median 4, range 1–10). Treatment modifications were required more frequently in cohort I than in cohort II (Table 2). Dose reductions became necessary after a median of three cycles of full-dose docetaxel and capecitabine in cohort I and three cycles of capecitabine and four cycles of full-dose docetaxel in cohort II, respectively. The total dose of chemotherapy that could be delivered was higher in cohort I. The main reasons for dose modifications were non-haematological toxicities. The majority of uncomplicated neutropenia was short lasting and did not result in a dose reduction. Calculating the planned dose of chemotherapy for the delivered number of cycles, in cohort I 88.2% of the planned dose of docetaxel and 83.7% of the planned dose of capecitabine could be administered compared with 94.2% of the planned dose of docetaxel and 92.4% of the planned dose of capecitabine administered in cohort II. The main reason for treatment termination was tumour progression (cohort I: 62.5%; cohort II: 78.4%). Toxicity (cohort I: 5%; cohort II: 5.9%) or withdrawal of consent (cohort I: 0%; cohort II: 2.0%) rarely lead to treatment termination.
Efficacy
A summary of confirmed response rates is presented in Table 3. Complete and partial responses were documented in 2 and 18 patients in cohort I, respectively, and in 0 and 12 patients in cohort II, respectively. The null hypothesis from the statistical design could be rejected in cohort I, but not in cohort II (see Figure 1). Overall response rates, the primary study end point, were 50.0% (n=20) in cohort I and 23.5% (n=12) in cohort II. The study was not powered or designed to compare the two cohorts. In an exploratory analysis, the observed difference was statistically significant (P=0.014) (Fisher's exact test, two sided).
Symptomatic improvement was evident in 27 of 37 (73.0%) patients with baseline symptoms in cohort I and 27 of 40 (67.5%) patients in cohort II.
Kaplan–Meier curves for time to tumour progression and overall survival are shown in Figure 2. Median time to tumour progression was 5.6 months (95% confidence intervals 3.9–7.3) in cohort I and 3.7 months (95% confidence intervals (CI) 2.5–5.0) in cohort II. Median overall survival was 10.1 months (95% CI: 7.3–13.0) in cohort I and 7.2 months (95% CI: 4.1–10.2) in cohort II. Exploratory statistical analysis of the observed differences in time to tumour progression (P=0.10, log-rank test; hazard ratio=0.71; 95% CI: 0.47–1.08) and overall survival (P=0.20, log-rank test; hazard ratio=0.75; 95% CI: 0.48–1.16) were not significant.

Time to tumour progression (A) and overall survival (B) in cohort I (higher dose, n=40) and cohort II (lower dose, n=51) treated with docetaxel and capecitabine. (A) Median time to tumour progression: cohort I, 5.6 months (95% CI: 3.9–7.3), cohort II, 3.7 months (95% CI: 2.5–5.0), P=0.10 (log-rank test). (B) Median overall survival: cohort I, 10.1 months (95% CI: 7.3–13.0), cohort II, 7.2 months (95% CI: 4.1–10.2), P=0.20 (log-rank test).
Safety
All patients were evaluable for safety. A summary of all adverse events by grade is provided in Table 4. All toxicities occurred more frequently in cohort I than in cohort II.
Non-haematological adverse events reported most frequently with the docetaxel–capecitabine regimen were asthenia, alopecia, nausea, hand–foot syndrome and diarrhoea. Regardless of the doses of capecitabine and docetaxel administered, these events were mostly mild to moderate in severity (grades 1 and 2), and grade 3 events were manageable (Table 4). Grade 4 non-haematological events were documented in a total of four patients: cohort I, vomiting (n=1) and pulmonary embolism (n=1); cohort II, cardiac arrhythmia (n=1) and pulmonary oedema (fluid retention) (n=1). One patient in cohort I died at home after the first cycle of chemotherapy due to a pulmonary embolism (grade 5 event).
Neutropenia and leukopenia were the most frequently reported haematological toxicity (Table 4). While both thrombocytopenia and anaemia were generally grade 1 or 2 events, grade 3/4 neutropenia and leukopenia were common in both patient cohorts. Grade 3 and 4 neutropenia was documented in 8 (20.0%) and 13 (32.5%) patients in cohort I, respectively, and in 4 (7.8%) and 8 (15.7%) patients in cohort II, respectively.
Discussion
Several modified forms of DCF have been tested in an attempt to improve the toxicity profile of this regimen (Park et al, 2005; Lorenzen et al, 2007; Al-Batran et al, 2008; Shah et al, 2010; Tebbutt et al, 2010). Only the US trial (Shah et al, 2010) included a reference regimen as comparator and could demonstrate a promising improvement of tolerability and efficacy by a modified DCF. In a previous phase II study, we demonstrated that docetaxel plus infused 5-fluorouracil had similar efficacy to the standard ECF regimen in the first-line treatment of patients with advanced gastric cancer (Thuss-Patience et al, 2005). As substitution of oral capecitabine for 5-fluorouracil is now accepted in patients with advanced gastric cancer (Cunningham et al, 2008; Kang et al, 2009), docetaxel and capecitabine is a logical combination in this setting. Several phase II studies have tested capecitabine in combination with docetaxel as first-line therapy in patients with advanced gastric cancer (Park et al, 2004; Chun et al, 2005; Kim et al, 2005; Giordano et al, 2006; Orditura et al, 2006; Tebbutt et al, 2010), but none has studied the effects of different doses.
In this study, we tested two different doses of docetaxel–capecitabine in sequential cohorts of patients with inoperable, locally advanced or metastatic gastric cancer. As it is not a randomised trial, by principle some bias cannot be ruled out. To minimise bias, consecutive patients were included. The two cohorts had similar baseline characteristics; the mild imbalance between gastro-oesophageal and gastric body cancer is not expected to influence primary end points, as published previously (Chau et al, 2009). A very promising response rate of 50.0% was observed in patients treated with higher doses of docetaxel–capecitabine (docetaxel 75 mg m−2 on day 1 plus capecitabine 2000 mg m−2 on days 1–14 every 3 weeks) compared with patients treated with lower doses (23.5%). Although the study was not designed to support a statistical comparison of the two patient cohorts, an exploratory analysis showed that there was a significant difference in favour of the higher dose regimen in terms of overall response rate. Time to tumour progression and overall survival were also both extended by approximately 2 months with the higher dose regimen compared with the lower dose regimen. These findings are mature as nearly all events had occurred at the time of analysis.
The toxicity profile of the docetaxel–capecitabine regimen in our study was characterised by mild-to-moderate non-haematological adverse events. Leukopenia/neutropenia was the most frequent haematological adverse event; grade 3/4 events were reported in 30–50% of patients depending on the docetaxel dose used. This profile of events is in keeping with other phase II studies of the docetaxel–capecitabine regimen (Park et al, 2004; Chun et al, 2005; Kim et al, 2005). Our data suggest that docetaxel–capecitabine at both dose levels investigated can be given safely in an ambulatory setting without unexpected toxicity. The lower dose regimen had an improved toxicity profile and required fewer dose reductions. Despite more dose reductions during the course of treatment, the higher dose regimen (cohort I) was also tolerable. A median of three cycles could be administered at full dose before dose reductions became necessary (Table 2).
To our knowledge, this is the first study that allows a comparison of two dose levels of the same regimen. Starting treatment with a higher dose of docetaxel and capecitabine is significantly more effective than starting with a moderate dose, even though many more dose reductions were necessary with the higher starting dose. It is the first study showing dose-dependent efficacy of docetaxel and capecitabine in gastric cancer.
This trial evaluates two different palliative strategies: strategy 1 – treat aggressively with a high-dose regimen and reduce doses as necessary; or strategy 2 – start cautiously with a less aggressive form of chemotherapy. On the basis of our findings, we suggest that a high-dose strategy, with dose reductions as needed, is more effective than a conservative approach. As the median age was only 62 years and only a minority of patients were 70 years and older (cohort I, 11 patients; cohort II, 13 patients), our trial unfortunately cannot answer the question whether this strategy also applies to elderly patients. Al-Batran et al (2010) could demonstrate that elderly patients also tolerate intense chemotherapy and benefit which would support this strategy. Further support for high starting doses with dose reductions as needed comes from the study of O’Shaughnessy et al (2002) in patients with metastatic breast cancer. In this study, it was demonstrated that the efficacy of docetaxel–capecitabine was not compromised by dose reductions required for the management of toxicity.
An alternate strategy might be to start at lower doses and escalate if possible. So far no data support this strategy in gastric cancer. In our view, it seems questionable whether similarly high treatment exposure can be achieved in the first cycles to result in best possible efficacy, but this needs to be investigated.
Several different regimens combining capecitabine and docetaxel have been tested in phase II trials as first-line therapy in patients with advanced gastric cancer (Table 5). Response rates in these studies ranged from 26 to 60%, median time to disease progression/progression-free survival from 4.4 to 5.2 months and median overall survival from 8.4 to 12.0 months. Lower doses of docetaxel administered weekly seem to be less promising in terms of efficacy compared with a 3-weekly administration. The efficacy of our higher dose regimen is consistent with these data, whereas our lower dose regimen is among the least active of those described (Table 5). The rates of neutropenia reported in these trials were highly variable, influenced by the dose and scheduling of docetaxel (Tebbutt et al, 2010).
The patients in our study were assigned to sequential cohorts rather than randomised to treatment. Although the baseline characteristics of the two cohorts were generally similar, the study should be viewed with this limitation in mind. The sample size was determined for each cohort separately rather than for a comparison of treatments; therefore, the statistical comparisons in the present report are exploratory only.
In conclusion, docetaxel–capecitabine is an active first-line regimen in patients with advanced gastric cancer, which can be given on an outpatient basis. We propose that it may be better to start docetaxel–capecitabine at a higher dose, with dose reductions as needed, rather than initiating therapy with lower doses to avoid dose reductions. It is a possible hypothesis that this strategy may be relevant for all first-line chemotherapy regimens in patients with advanced gastric cancer, although this requires confirmation in prospective trials.
References
Al-Batran SE, Hartmann JT, Hofheinz R, Homann N, Rethwisch V, Probst S, Stoehlmacher J, Clemens MR, Mahlberg R, Fritz M, Seipelt G, Sievert M, Pauligk C, Atmaca A, Jäger E (2008) Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol 19: 1882–1887
Al-Batran S, Hohmann N, Hartmann JT, Moehler MH, Pauligk C, Probst S, Rethwisch V, Prasnikar N, Stoehlmacher J, Jaeger E (2010) 5-fluorouracil, leucovorin, and oxaliplatin with or without docetaxel in elderly (65 years or older) patients with esophagogastric cancer: FLOT65+ trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO). J Clin Oncol 28 (Suppl): 15s (abstract 4013)
Chau I, Norman AR, Cunningham D, Oates J, Hawkins R, Iveson T, Nicolson M, Harper P, Seymour M, Hickish T (2009) The impact of primary tumour origins in patients with advanced oesophageal, oesophago-gastric junction and gastric adenocarcinoma – individual patient data from 1775 patients in four randomised controlled trials. Ann Oncol 20: 885–891
Chun JH, Kim HK, Lee JS, Choi JY, Hwangbo B, Lee HG, Park SR, Choi IJ, Kim CG, Ryu KW, Kim YW, Lee JS, Bae JM (2005) Weekly docetaxel in combination with capecitabine in patients with metastatic gastric cancer. Am J Clin Oncol 28: 188–194
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman AR, Upper Gastrointestinal Clinical Studies Group of the National Cancer Research Institute of the United Kingdom (2008) Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 358: 36–46
Giordano KF, Jatoi A, Stella PJ, Foster N, Tschetter LK, Alberts SR, Dakhil SR, Mailliard JA, Flynn PJ, Nikcevich DA, North Central Cancer Treatment Group (2006) Docetaxel and capecitabine in patients with metastatic adenocarcinoma of the stomach and gastroesophageal junction: a phase II study from the North Central Cancer Treatment Group. Ann Oncol 17: 652–656
Kamangar F, Dores GM, Anderson WF (2006) Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol 24: 2137–2150
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L, Philco-Salas M, Suarez T, Santamaria J, Forster G, McCloud PI (2009) Capecitabine/cisplatin vs 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 20: 666–673
Kim JG, Sohn SK, Kim DH, Baek JH, Sung WJ, Park JY, Kim TB, Jung HY, Yu W, Lee KB (2005) Phase II study of docetaxel and capecitabine in patients with metastatic or recurrent gastric cancer. Oncology 68: 190–195
Kim NK, Park YS, Heo DS, Suh C, Kim SY, Park KC, Kang YK, Shin DB, Kim HT, Kim HJ (1993) A phase III randomized study of 5-fluorouracil and cisplatin vs 5-fluorouracil, doxorubicin, and mitomycin C vs 5-fluorouracil alone in the treatment of advanced gastric cancer. Cancer 71: 3813–3818
Lee KS, Lee HY, Park EK, Jang JS, Lee SJ (2008) A phase II study of leucovorin, 5-FU and docetaxel combination chemotherapy in patients with inoperable or postoperative relapsed gastric cancer. Cancer Res Treat 40 (1): 11–15
Lo SS, Khorana AA, Javle M, Simon S, Kiefer G, Rajasenan K, Wang H, Hantel A, Shayne M, Hwang J, Schmotzer A, Ramanathan RK (2010) A phase II study of weekly docetaxel in combination with capecitabine in advanced gastric and gastroesophageal adenocarcinomas. Oncology 78: 125–129
Lorenzen S, Duyster J, Lersch C, von Delius S, Hennig M, Bredenkamp R, Peschel C, Lordick F (2005) Capecitabine plus docetaxel every 3 weeks in first- and second-line metastatic oesophageal cancer: final results of a phase II trial. Br J Cancer 92: 2129–2133
Lorenzen S, Hentrich M, Haberl C, Heinemann V, Schuster T, Seroneit T, Roethling N, Peschel C, Lordick F (2007) Split-dose docetaxel, cisplatin and leucovorin/fluorouracil as first-line therapy in advanced gastric cancer and adenocarcinoma of the gastroesophageal junction: results of a phase II trial. Ann Oncol 18: 1673–1679
Ohtsu A, Shimada Y, Shirao K, Boku N, Hyodo I, Saito H, Yamamichi N, Miyata Y, Ikeda N, Yamamoto S, Fukuda H, Yoshida S (2003) Randomized phase III trial of 5-fluorouracil alone vs 5-fluorouracil plus cisplatin vs uracil and tegafur plus mitomycin C in patients with unresectable advanced gastric cancer: The Japan Clinical Oncology Group Study (JCOG9205). J Clin Oncol 21: 54–59
Orditura M, Martinelli E, Galizia G, Carlomagno C, Aurilio G, Vecchione L, Lieto E, De Placido S, Catalano G, Ciardiello F, De Vita F (2006) Weekly docetaxel and capecitabine is not effective in the treatment of advanced gastric cancer: a phase II study. Ann Oncol 17: 1529–1532
O’Shaughnessy J, Miles D, Vukelja S, Moiseyenko V, Ayoub JP, Cervantes G, Fumoleau P, Jones S, Lui WY, Mauriac L, Twelves C, Van-Hazel G, Verma S, Leonard R (2002) Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol 20: 2812–2823
Park SR, Chun JH, Kim YW, Lee JH, Choi IJ, Kim CG, Lee JS, Bae JM, Kim HK (2005) Phase II study of low dose docetaxel/fluorouracil/cisplatin in metastatic gastric carcinoma. Am J Clin Oncol 28: 433–438
Park YH, Ryoo BY, Choi SJ, Kim HT (2004) A phase II study of capecitabine and docetaxel combination chemotherapy in patients with advanced gastric cancer. Br J Cancer 90: 1329–1333
Parkin DM, Pisani P, Ferlay J (1999) Global cancer statistics. CA Cancer J Clin 49: 33–64
Rosati G, Bilancia D, Germano D, Dinota A, Romano R, Reggiardo G, Manzione L (2007) Reduced dose intensity of docetaxel plus capecitabine as second-line palliative chemotherapy in patients with metastatic gastric cancer: a phase II study. Ann Oncol 18 (Suppl 6): vi128–vi132
Ross P, Nicolson M, Cunningham D, Valle J, Seymour M, Harper P, Price T, Anderson H, Iveson T, Hickish T, Lofts F, Norman A (2002) Prospective randomized trial comparing mitomycin, cisplatin, and protracted venous-infusion fluorouracil (PVI 5-FU) with epirubicin, cisplatin, and PVI 5-FU in advanced esophagogastric cancer. J Clin Oncol 20: 1996–2004
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10: 1–10
Shah MA, Shibata S, Stoller RG, Kemeny M, Ritch PS, Krishnamurthi SS, Su YB, Janjigian YY, Capanu M, Kelsen DP (2010) Random assignment multicenter phase II study of modified docetaxel, cisplatin, fluorouracil (mDCF) vs DCF with growth factor support (GCSF) in metastatic gastroesophageal adenocarcinoma (GE). J Clin Oncol 28 (Suppl): 15s (abstract 4014)
Tebbutt NC, Cummins MM, Sourjina T, Strickland A, Van Hazel G, Ganju V, Gibbs D, Stockler M, Gebski V, Zalcberg J, Australasian Gastro-Intestinal Trials Group (2010) Randomised, non-comparative phase II study of weekly docetaxel with cisplatin and 5-fluorouracil or with capecitabine in oesophagogastric cancer: the AGITG ATTAX trial. Br J Cancer 102: 475–481
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van-Glabbeke M, van-Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216
Thuss-Patience PC, Kretzschmar A, Repp M, Kingreen D, Hennesser D, Micheel S, Pink D, Scholz C, Dörken B, Reichardt P (2005) Docetaxel and continuous-infusion fluorouracil vs epirubicin, cisplatin, and fluorouracil for advanced gastric adenocarcinoma: a randomized phase II study. J Clin Oncol 23: 494–501
Van Cutsem E, Moiseyenko VM, Tjulandin SA, Majilis J, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, Risse ML, Ajani JA, V325 Study Group (2006) Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 study group. J Clin Oncol 24: 4991–4997
Vanhoefer U, Rougier P, Wilke H, Ducreux MP, Lacave AJ, Van Cutsem E, Planker M, Santos JG, Piedbois P, Paillot B, Bodenstein H, Schmoll HJ, Bleiberg H, Nordlinger B, Couvreur ML, Baron B, Wils JA (2000) Final result of a randomized phase III trial of sequential high-dose methotrexate, fluorouracil, and doxorubicin, vs etoposide, leucovorin, and fluorouracil vs infusional fluorouracil and cisplatin in advanced gastric cancer: a trial of the European Organization for Research and Treatment of Cancer Gastrointestinal Tract Cooperative Group. J Clin Oncol 81: 2648–2657
Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, Fleig WE (2010) Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev 3: CD004064
Webb A, Cunningham D, Scarffe H, Harper P, Norman A, Joffe JK, Hughes M, Mansi J, Findlay M, Hill A, Oates J, Nicolson M, Hickish T, O’Brien M, Iveson T, Watson M, Underhill C, Wardley A, Meehan M (1997) Randomized trial comparing epirubicin, cisplatin, and fluorouracil, vs fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol 15: 261–267
Acknowledgements
The study was supported by a research grant from Roche and Sanofi-Aventis. We thank Ms Bettina Lebedinzew and Ms Simone Micheel for their devoted work at the study secretariat and Mr Lee Miller (Miller Medical Communications, UK) for his support in preparing the manuscript. We thank the AIO steering committee (gastro-oesophageal cancer division) for their very fruitful discussions, and Dr Rosemarie Kuhl (Sanofi-Aventis), Ms Sabine Tondar (Roche) and Dr Silke Günther (Roche) for their excellent cooperation. Our special thanks go to all patients who participated in this trial.
Author information
Authors and Affiliations
Corresponding author
Additional information
The study was presented in part at the annual meeting of the American Society of Clinical Oncology in 2008.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Thuss-Patience, P., Kretzschmar, A., Dogan, Y. et al. Docetaxel and capecitabine for advanced gastric cancer: investigating dose-dependent efficacy in two patient cohorts. Br J Cancer 105, 505–512 (2011). https://doi.org/10.1038/bjc.2011.278
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.278
