
Abstract
PRDM1/Blimp-1 is a tumor suppressor gene in the activated B-cell subtype of diffuse large B-cell lymphomas. Its inactivation contributes to pathogenesis in this setting by impairing terminal B-cell differentiation induced by constitutive nuclear factor-κB activation. The role of PRDM1 in Burkitt lymphoma (BL) lymphomagenesis is not known. Here we identified hypermethylation of the promoter region and exon 1 of PRDM1 in all six Epstein–Barr virus (EBV)-positive BL cell lines and 12 of 23 (52%) primary EBV-positive BL or BL-related cases examined, but in none of the EBV-negative BL cell lines or primary tumors that we assessed, implying a tumor suppressor role for PRDM1 specifically in EBV-associated BL. A direct induction of PRDM1 hypermethylation by EBV is unlikely, as PRDM1 hypermethylation was not observed in EBV-immortalized B lymphoblastoid cell lines. Treatment of EBV-positive BL cells with 5′ azacytidine resulted in PRDM1 induction associated with PRDM1 demethylation, consistent with transcriptional silencing of PRDM1 as a result of DNA methylation. Overexpression of PRDM1 in EBV-positive BL cell lines resulted in cell cycle arrest. Our results expand the spectrum of lymphoid malignancies in which PRDM1 may have a tumor suppressor role and identify an epigenetic event that likely contributes to the pathogenesis of BL.
Similar content being viewed by others


Introduction
PRDM1/Blimp-1 is a DNA-binding, PR (positive regulatory) domain-containing transcription repressor that has a key role in the terminal differentiation of B cells, as well as in the homeostatic maintenance of T cells.1 Its function in regulating differentiation, activation and homeostasis also extend to other cell types.2 A role for PRDM1 in lymphoma biology was first implicated when inactivating nonsense mutations, the vast majority of which associated with allelic deletions, were found to be present in almost a quarter of diffuse large B-cell lymphoma (DLBCL) of the non-germinal center (GC) B-cell/activated B-cell subtype.3,4 More recently, the tumor suppressor activity of PRDM1/Blimp-1 in DLBCL has been confirmed in two mouse models.5,6 PRDM1 expression or activity is also downregulated by other mechanisms in non-GC B-cell type of DLBCL, including missense mutations, biallelic mutations and constitutively active translocated BCL6 (a potent repressor of PRDM1 transcription).5 Posttranscriptional downregulation of PRDM1 by microRNAs has also been implicated.7 These findings strongly implicate an important role of impairment of PRDM1-mediated terminal B-cell differentiation in the pathogenesis of the activated B-cell/non-GC B-cell subtype of DLBCL, which is characterized by constitutive activation of nuclear factor-κB (NF-κB),8 a strong PRDM1/Blimp-1 inducer.1 Recently, PRDM1 was also implicated as a tumor suppressor gene in aggressive natural killer cell lymphomas and anaplastic large-cell lymphoma.9, 10, 11
Burkitt lymphoma (BL) is an aggressive B-cell lymphoma characterized by reciprocal translocations between immunoglobulin gene loci, mostly IgH, and the c-myc locus. BL consists of three clinical subtypes: endemic, sporadic and immunodeficiency associated. BLs frequently harbor latent Epstein–Barr virus (EBV) infection, with 100% EBV positivity in the endemic form, ~30% within sporadic cases and 25–40% in immunodeficiency-associated BL.12 Transcriptome profiling identified a distinct gene expression signature for BL closely related to early GC B cells, with slight but significant differences among subsets.13 It has been previously suggested that unlike EBV-negative BL, EBV-positive BL may be derived from a precursor cell corresponding to the post-GC/memory B-cell stage of differentiation based on the pattern of IgH somatic hypermutation.14 However, data generated from gene expression profiling is more consistent with a GC B-cell origin, regardless of EBV status.
BL cells typically express BCL6 and CD10, lack BCL2 and are consistently negative for PRDM1.15 A role for PRDM1 inactivation in BL is currently not known. However, several observations suggest that PRDM1 is a target for inactivation in BL. First, PRDM1 is one of the targets for microRNA-127, which is overexpressed in EBV-positive BL, suggesting that its downregulation may be important for EBV-associated BL pathogenesis.16 Second, comparison of the gene expression profiles of endemic (EBV-positive) and sporadic (generally EBV-negative) BLs revealed increased activity in the NF-κB pathway, among several others, in the former,13 raising the possibility for the need to dampen PRDM1 activity to prevent NF-κB-induced terminal differentiation in a subset of BLs. Lastly, expression of PRDM1, which can lead to virus replication and cell death in EBV-infected GC B cells, is downregulated by EBV-encoded latent membrane protein-1 (LMP1).17 Collectively, these observations suggest that, in effect, PRDM1 may have a tumor suppressor role in EBV-related BL.
Here we demonstrate frequent hypermethylation within the 5′ region of the PRDM1 gene that is exclusive to the EBV-positive subset of BL, suggesting that this epigenetic alteration may have a unique role in the pathogenesis of EBV-associated BL. Our findings thus support the notion for a tumor suppressor role of PRDM1 in a widening spectrum of lymphoid tumors, and the potential importance of PRDM1 inactivation in EBV-associated malignancies.
Materials and methods
Cell lines
All BL cell lines were cultured in RPMI-1640 medium supplemented with L-glutamine and 10% heat-inactivated fetal bovine serum (Atlanta Biologicals Inc., Lawrenceville, GA, USA).
Patient tissue samples
Formalin-fixed, paraffin-embedded archival tissue of BL cases were selected from the pathology archives of Weill Cornell Medical College and Consultoria em Patologia (Sao Paulo, Brazil) according to the protocols approved by the Institutional Review Board. All samples were reviewed and classified according to the World Health Organization criteria.12
Normal B-cell subsets
GC, naive and memory B cells were sorted from B cells of reactive tonsils by flow cytometry as described previously.7
Methylation analysis
A CG island was predicted to exist in the 5′ region of PRDM1 by MethPrimer.18 Promoter and exon 1 methylation in PRDM1 was assessed by bisulfite sequencing. DNAs from BL cell lines or formalin-fixed, paraffin-embedded primary BL cases were modified with bisulfite using the Epitect Bisulfite Kit (Qiagen) according to the manufacturer’s protocol, and amplified using primers that did not contain CpG. Primers were designed using the MethPrimer program18 and had the following nucleotide sequences: A (forward), 5′-TTTTTGTATTTGGGGATTTGAGTT-3′; B (reverse), 5′-AAAATCTTCTTACTTCCCTTTAAAAACA-3′; C (forward), 5′-GTGTTTTTAAAGGGAAGTAAGAAGATTT-3′; D (reverse), 5′-TAACTTCCCCTCCCTACTTAAAATT-3′; E (forward), 5′-TTTTAAGTAGGGAGGGGAAGTTAGA-3′; F (reverse), 5′-CAAACAAATATCCAACATCTAAAAAAAA-3′.
For the analysis of methylation of DNA isolated from cell suspension, primers A and F were used to generate an amplicon of 601 bp that contains 41 CpG dinuleotide pairs (chr6:106 533 846–106 534 446, February 2009 build). For the methylation analysis of bisulfite-modified DNA isolated from formalin-fixed, paraffin-embedded tissues, three primer pairs were used, A and B, C and D and E and F, to divide the larger amplicon into three separate, partially overlapping, shorter segments to facilitate PCR amplification of DNA extracted from formalin-fixed, paraffin-embedded tissues. These primers generated amplicons of 192, 195 and 266 bp (including primers), respectively. Similar to the 601 amplicon, the combination of these three smaller amplicons also contained the same 41 CpG sites. Amplified DNA was ligated into pGEM-T (Promega, Madison, WI, USA) and the DNA sequences of at least 10–12 clones per bisulfite reaction were determined by Sanger sequencing and analyzed with a BiQ analyzer (Max-Planck-Institut Informatik, Saarbrücken, Germany).19
Quantitative reverse transcriptase-PCR
Quantitative detection of PRDM1α mRNA was performed as described previously.20
Immunohistochemistry
Immunohistochemical staining of LMP1 (1:50, clone CS.1–4; Dako, Carpinteria, CA, USA), EBNA2 (1:50, clone PE2; Dako), BCL6 (1:10, clone PG-B6p; Dako), PRDM1 (1:50, clone 3H2E8; Santa Cruz Biotechnology, Dallas, TX, USA) and MUM1/IRF4 (1:100, clone MUM1p; Dako) were accomplished using the Bond III Autostainer (Leica Microsystems, Buffalo Grove, IL, USA). Formalin-fixed, paraffin-embedded tissues or cell block sections were first baked and deparaffinized. Antigens were then retrieved by heating the slides at 37 °C in Bond Enzyme solution (Leica Microsystems) for 10 min (for LMP1), and at 99–100 °C in Bond Epitope Retrieval Solution 2 for 20 (for EBNA2, PRDM1 and MUM1) or 30 min (for BCL6). Sections were then incubated sequentially with the endogenous peroxidase block, primary antibody, postprimary (equivalent to secondary antibody), polymer (equivalent to tertiary antibody), diaminobenzidine and hematoxylin for 5, 25, 15, 25, 10 and 5 min, respectively. Bond Polymer Define Detection (Leica Microsystems) was used for EBNA2 and MUM1, and Bond Polymer Refine Detection (Leica Microsystems) was used for LMP1, BCL6 and PRDM1. Finally, the stained slides were dehydrated and mounted in Cytoseal XYL (Richard-Allan Scientific, Kalamazoo, MI, USA).
Statistical analysis
P-values were calculated by Student’s t-test and Mann–Whitney U-test using the JMP software (SAS Institute Inc., Cary, NC, USA).
Treatment of BL cell lines with 5′ azacytidine
EBV-positive and -negative BL cell lines were incubated with 10 μM of dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St Louis, MO, USA) or 5′ azacytidine (AZA) (Sigma-Aldrich; no. A3656) for 4 days. PRDM1 expressions in DMSO- and AZA-treated cells were determined by quantitative reverse transcriptase (RT)-PCR.
Plasmid construction
The PRDM1-PMSCV-PIG expression plasmid was generated by PCR amplification using cDNA prepared from the myeloma cell line U266 as a template, PRDM1-F: 5′-AAGGTCGACATGTTGGATATTTGCTTGGAA-3′ as a forward primer and PRDM-R: 5′-GCCGAATTCTTAAGGATCCATTGGTTCA-3′ as a reverse primer. The PCR fragment was then gel purified and double digested with XhoI and EcoRI before subcloning into the PSMCV-PIG vector (Addgene, Cambridge, MA, USA). The sequence of the insert was confirmed by Sanger sequencing and by comparison with the PRDM1 sequence in the database (NM_001198.3).
Transfection
BL cells were transfected with the indicated amount of PRDM1-PIG or PMSCV-PIG empty vector using Neon transfection system (Life Technologies Inc., Grand Island, NY, USA) with program no. 16 according to the instruction manual. Mock transfection was also included as a negative control.
Western blotting
A total of 5 × 106 BL cells were transfected with 10 μg of PRMD1-PIG or PMSCV-PIG plasmids. At 48 h after transfection, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% sodium dodecyl sulfate, 2 mM EDTA, 1x protease inhibitor cocktail II and III (Calbiochem, San Diego, CA, USA) and 1x phosphatase inhibitor (Sigma-Aldrich)) to collect whole-cell extracts. After gel electrophoresis and transfer, the membranes were then probed with rabbit monoclonal antibody against PRDM1 (clone C14A4; Cell Signaling Technology, Danvers, MA, USA) at 4 °C overnight, incubated with IRDye 680 goat anti-rabbit IgG (Li-Cor, Lincoln, NE, USA) at room temperature for 1 h and were scanned with Odyssey imager (Li-Cor). Lamin B (Santa Cruz Biotechnology) was loaded as a normalization control.
Apoptosis and cell cycle analysis
A total of 0.5 × 106 cells were transfected with 1 μg of the above indicated plasmids, and the number of dead cells was measured at 48 h time point by flow cytometry using the Annexin V/7ADD Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA); bromodeoxyuridine (BrdU) incorporation was analyzed using the V450 BrdU Flow Kit (BD Biosciences) according to the manufacturer’s instruction. In brief, cells were incubated with 10 μM BrdU at 37 °C for 2 h and stained with V450-conjugated anti-BrdU antibody (BD Biosciences) for 30 min at room temperature under the dark. Cells were then resuspended in 400 μl staining buffer and subjected to LSRII flow cytometer for analysis. The percentage of cell cycle distribution was measured by FlowJo software (Tree Star Inc., Ashland, OR, USA). The experiments were repeated three separate times.
Results
Hypermethylation of PRDM1 promoter and exon 1 in EBV-positive BL cell lines and primary BL cases
Sequencing of the PRDM1 coding region in BL cell lines did not reveal somatic mutations (Supplementary Table 1). To investigate the possibility of epigenetic inactivation of PRDM1 in BL, we first performed bisulfite sequencing to assess the methylation status of the 5′ end of PRDM1 (−325 to +224), which encompasses a CpG island that includes 41 CpG dinucleotides, in six EBV-positive and two EBV-negative BL cell lines (Figure 1). Four of the six EBV-positive BL cell lines (Akata, Kem I, Mutu I and RaeI) support the EBV latency I transcription program and two (Daudi and P3HR1) the Wp-restricted program.21 PRDM1 hypermethylation was detected in all six EBV-positive BL cell lines (Figure 2a). By contrast, among EBV-positive lymphoblastoid cell lines (LCLs), there was little or no methylation, suggesting that EBV itself does not induce methylation of PRDM1. Likewise, PRDM1 hypermethylation was not observed within EBV-negative BL cell lines (Ramos, DG-75) or in normal B-cell subsets, including GC, memory and naive B cells.

CG-rich region at the 5′ end of the PRDM1 gene. (a) The 5′ region of the PRDM1 gene is predicted to contain a CG island spanning the promoter region and exon 1. The genomic region analyzed by bisulfite sequencing (−325 to +224) is indicated. The filled region indicates the open reading frame; the bent arrow denotes the transcription start site. (b) The sequence at the 5′ end of PRDM1, including the region −325 to +224 analyzed by bisulfite sequencing, is depicted. The three portions of sequence that were separately analyzed in formalin-fixed, paraffin-embedded primary BLs are highlighted in gray. The 41 CpG dinucleotides interrogated are numbered within the sequence. The boundaries of the predicted CpG island are indicated by horizontal arrows.

EBV-positive BL cell lines and primary tumors are frequently methylated in the PRDM1 gene. (a) Bisulfite sequencing of the PRDM1 gene was performed on eight EBV-positive BL cell lines, two EBV-negative BL cell lines, five LCL and six normal B-cell subets (two naive, two memory and two GC B cells). PRDM1 hypermethyation was observed exclusively in EBV-positive cell lines analyzed. (b) The distribution and PRDM1 methylation status of the analyzed BL cases are illustrated by pie charts. A total of 44 BLs or BL-related lymphomas were analyzed, of which 23 were EBV-positive and 21 were EBV-negative. They were comprised of 30 non-HIV BL, 9 HIV-related BL and 5 BCL, U. PRDM1 hypermethylation was seen in 12 of 23 EBV-positive BL/BCL, U, but none of the 21 EBV-negative BL/BCL, U cases (P<0.05).
We next investigated whether similar PRDM1 hypermethylation can be seen in primary BL cases. We initially collected a cohort of 42 BL cases from Brazil, and from the United States a cohort of 27 BL and 5 unclassifiable B-cell lymphomas (BCL, Us) with features intermediate between BL and DLBCL.22 Among these, 17 (10 EBV-positive, 7 EBV-negative) of the 42 BLs from Brazil and 27 (13 EBV-positive, 14 EBV-negative) of 32 BL/BCL, U from the United States yielded interpretable results. The BL subtype (non-HIV-related vs HIV-related), patient age and EBV status of these are summarized in Table 1. Of note, the age at diagnosis for the EBV-positive, non-HIV BL subgroup (median=6) was lower than that for the EBV-negative, non-HIV BL subgroup (median=14; P<0.05, Mann–Whitney test).
PRDM1 promoter/exon 1 hypermethylation was identified in 11 of 22 (50%) EBV-positive BL and 1 of 1 (100%) EBV-positive BCL, U, but in none of the 17 EBV-negative BL and 4 EBV-negative BCL, U cases (P=0.0006 for BL only (n=39); P=0.0001 for BL and BCL, U (n=44); Figure 2b). Among the 12 PRDM1-hypermethylated, EBV-positive cases, 8 were non-HIV BL, 3 were HIV-related BL and 1 was BCL, U. Seven of the 8 PRDM1-hypermethylated, non-HIV BL belonged to the Brazil cohort and 1 belonged to the US cohort. Seventy percent (7 of 10) of the Brazilian non-HIV, EBV-positive BL cases had hypermethylated PRDM1, whereas 20% (1 of 5) of the US non-HIV, EBV-positive BL cases had hypermethylated PRDM1 (P=0.06). Fifty percent (4 of 8) of the HIV-related, EBV-positive BL or BCL, U cases harbor hypermethylated PRDM1. Among the EBV-positive cases, there was no significant difference in MUM1/IRF4 positivity (as determined by immunohistochemistry) and patient age between those with hypermethylated or unmethylated PRDM1 (Table 1).
Methylation results for each of the 41 CpGs from the distal promoter through the first exon are summarized in Figure 3. On average, the percentage of methylation across the interrogated CpGs ranged from about 40–50 to 100% across or within some regions of the assessed domain in the EBV-positive BL cell lines. The extent of methylation in primary EBV-positive BL cases overall was lower and more variable compared with the BL cell lines, with roughly half exhibiting the hypomethylation noted for normal B cells, LCLs and primary cases of EBV-negative lymphoma. The rest of the EBV-positive cases demonstrated various overlapping patterns of local or more global hypermethylation not seen in the other EBV-postive cases or EBV-negative cases showing PRDM1 hypomethylation. In the majority of these cases, hypermethylation appeared subclonal, with <50% of methylation in the CpGs.

Methylation analysis of PRDM1 in BL cell lines and primary tumors. The percentage of methylation determined by bisulfite cloning and sequencing at each of the 41 CpG dinucleotides from −325 and +224 is summarized for all BL cell lines and primary cases, as well as for LCLs and normal B-cell subsets. BL cell lines and primary cases demonstrating PRDM1 hypermethylation, all of which were EBV-positive, are highlighted in gray. Significant hypermethylation was not observed in EBV-positive LCLs, normal B cells or EBV-negative BL cell lines or primary cases. Cases E3 and F1 were diagnosed as BCL, U, with features intermediate between BL and DLBCL (BCL, U). The remaining primary cases were BLs.
PRDM1 hypermethylation is associated with low PRDM1 expression in EBV-positive BL
Real-time RT-PCR analysis of the BL cell lines showed that they uniformly expressed very low levels of PRDM1α mRNA regardless of PRDM1 methylation status (Figure 4). In line with these results, BLs have been shown by immunohistochemistry to consistently lack PRDM1.15 We also found our cohorts of EBV-positive BLs to be negative for PRDM1 by immunoperoxidase staining (data not shown). In contrast, LCLs expressed variable but much higher (>10- to ~1000-fold) levels of PRDM1α mRNA (Figure 4). Differences in the levels of BCL6, a repressor of PRDM1 transcription,23 EBV latency program and PRDM1 methylation status may explain these distinct differences in PRDM1 expression between EBV-positive BLs and LCLs. In the latter, the presence of both positive regulatory signals, most likely related to the latency III EBV gene expression program, and the absence of inhibitory mechanisms (absence of BCL6 and PRDM1 hypermethylation) are conducive to PRDM1 induction. In contrast, BCL6 expression and PRDM1 hypermethylation in the former may serve as alternative mechanisms to inhibit PRDM1 expression in the setting of PRDM1-inducing signals during latency I (see also Discussion).

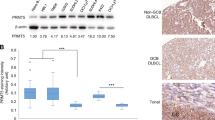
Expression of PRDM1α mRNA in BL and LCL cell lines. PRDM1α mRNA levels in eight BL cell lines (six EBV-positive) and five LCLs were measured by real-time RT-PCR. Values are relative to the mRNA level in the myeloma cell line U266 (set as 100). BL cell lines that showed hypermethylation at the 5′ region are indicated. BCL6 status in these cell lines was determined by immunohistochemistry (data not shown).
AZA treatment results in the upregulation of PRDM1 expression in EBV-positive BL cells
We next treated EBV-positive BL cells with the DNA methyltransferase inhibitor AZA to test directly whether DNA methylation contributes to PRDM1 silencing in the context of BL. Treatment of EBV-positive BL cell lines with AZA (10 μM) for 4 days resulted in ~110- to >4000-fold induction of PRDM1α mRNA relative to DMSO-treated controls (Figure 5a). In contrast, the EBV-negative cell line DG-75 showed much lower extent of PRDM1 induction (~9-fold). To confirm demethylation in EBV-positive BL cells by AZA, we analyzed PRDM1 methylation in Mutu I by bisulfite sequencing and revealed a 30–40% reduction in CpG methylation across the promoter region in treated Mutu I cells (Figure 5b). In addition, we observed a conversion from a latency I gene expression program (in which EBNA1 is expressed) to a latency III-type program associated with the expression of LMPs and other EBNAs in addition to EBNA1,24,25 as well as a marked decrease in BCL6 levels (Figure 5c). In conjunction with the observation that PRDM1 mRNA is highly expressed in LCLs, these findings suggest that the increase in PRDM1 mRNA observed in the EBV-positive cell lines upon treatment with AZA may be a result of a combinatorial effect of PRDM1 demethylation, generation of PRDM1-inducing signals by one or more of the latency III EBV gene products and downregulation of BCL6.

AZA treatment of EBV-positive BL cells resulted in robust increase in PRDM1 mRNA. (a) EBV-positive and EBV-negative BL cell lines were treated with AZA and the levels of PRDM1α mRNA were measured by quantitative RT-PCR. The results were expressed relative to DMSO-treated BL cells (DMSO-treated =1), The mean values (±s.e.) from three independent experiments are shown. (b) Average methylation percentage of each of the 41 CpGs within the 5′ region of PRDM1 determined by bisulfite sequencing is shown for Mutu I. (c) Induction of EBV latency III-associated LMP1 and EBNA2, and decrease in BCL6 upon AZA treatment of Mutu I cells, determined by immunohistochemistry on cell block materials. Original magnification x400 with x40 objective lens. Microscope: Olympus BX 41; Camera: Olympus Q-COLOR3; software: QCapture, version 2.9.8.0 (Quantitative Imaging Corporation, Surrey, BC, Canada).
Ectopic expression of PRDM1 in EBV-positive cell lines inhibits cell cycle progression at the G1 to S phase
The presence of PRDM1 methylation in EBV-positive BL cells suggests that epigenetic repression of PRDM1 expression confers selective growth advantage to these cells. To investigate the biological consequence of overexpression of PRDM1 in EBV-positive cells, PRDM1 was overexpressed in two EBV-positive cell lines, Daudi and P3HR1, and the effects on cell cycle and apoptosis were examined. Increase in PRDM1 levels in these cell lines resulted in G0/G1 arrest, with no effect on apoptosis (Figures 6a and c). Relative to control transfectants, an increased percentage of GFP-positive cells were retained in the G0/G1 phase upon transient transfection with the PRDM1-expressing construct PRDM1-PIG (40.7% vs 28.7%, P=0.0035 for Daudi; 43.6% vs 30.2%, P=0.0144 for P3HR1). The percentage of GFP-positive cells were correspondingly decreased (33.1% vs 46.5%, P=0.0053 for Daudi; 38.4% vs 53.8%, P=0.0015 for P3HR1). These results suggest that inhibition of PRDM1 levels in EBV-positive BL cells may be beneficial to tumor growth by preventing cell cycle arrest at the G1-to-S phase transition.

PRDM1 induces cell cycle arrest but not apoptosis in BL cells. (a) BL cells (Daudi and P3HR1) were transfected with PRMD1-PIG or PMSCV-PIG plasmids. Western blotting for PRDM1 was performed 48 h posttransfection. Lamin B was included as a loading control. (b) Apoptosis of GFP-positive cells measured by Annexin V/7ADD at 48 h posttransfection. Error bars indicate the s.e.m. from three independent experiments. NS, not significant. (c) Cell cycle analysis of GFP-positive populations at 48 h posttransfection. Transfected BL cells were labeled with 10 μM BrdU for 2 h, followed by intracellular staining with BrdU and 7-amino-actinomycin D (7-AAD) and flow cytometric analysis. Percentages of cell cycle events at different phases were calculated using the FlowJo software and the graph was plotted using Prism 6 software (GraphPad, La Jolla, CA, USA). Error bars denote the s.e.m. from three independent experiments, and the P-values were indicated. A representative graph was shown.
Discussion
PRDM1 is a tumor suppressor gene in DLBCL of the activated B-cell type, and also in natural killer cell malignancies and anaplastic large-cell lymphoma.10,11,26 The findings here of frequent PRDM1 promoter and exon 1 hypermethylation in a subset of BL are novel and have not been previously described in B-cell lymphomas. They expand the spectrum of B-cell lymphomas beyond DLBCL in which PRDM1 likely has a tumor suppressor role, and supports the importance of impairment of PRDM1-mediated functions in the pathogenesis of some aggressive B-cell lymphomas. Recently, PRDM1 has been shown to be a target for downregulation by an overexpressed microRNA, miR-127, in EBV-positive BL.16 Our findings support an additional mechanism in dampening PRDM1 activity in EBV-positive BL. Hypermethylation of the PRDM1 gene has also recently been shown in natural killer cell malignancies.10,27 Interestingly, the region of hypermethylation in natural killer tumors is limited to the more distal promoter (−100 to −300), in contrast to the more widespread methylation extending from distal promoter through exon 1 in EBV-positive BL as shown here. The mechanistic origin of this difference in methylation and whether this may translate to significant differences in transcription remains to be investigated.
The observation that PRDM1 promoter and exon 1 hypermethylation is exclusively seen in the EBV-positive subset of BL implies a specific and unique contribution of this epigenetic alteration to EBV-positive BL pathogenesis. There appears to be a tendency (P=0.06) for PRDM1 hypermethylation to be present more frequently in the Brazilian non-HIV, EBV-positive cohort than in the US non-HIV, EBV-positive cohort. BL cases in Brazil, particularly the EBV-positive ones, may be more akin to endemic BL rather than sporadic BL because of their association with chronic schistosome infection. Interestingly, all EBV-positive BL cell lines examined in our study, which were derived from endemic BL tumors, exhibited PRDM1 hypermethylation. Thus, PRDM1 hypermethylation may be the preferential mechanism to downregulate PRDM1 expression in EBV-positive BL cases of the endemic subtype. HIV-related, EBV-positive cases may also demonstrate increased the frequency of PRDM1 methylation compared with the US sporadic EBV-positive BL cases; however, no statistical significance could be obtained because of the limited sample size. There was no correlation between PRDM1 hypermethylation status and MUM1/IRF4 expression in the EBV-positive BL cases. MUM1 and PRDM1 are normally coexpressed in GC B cells destined to undergo plasma cell differentiation.28 A positive correlation would have supported the idea that PRDM1 hypermethylation contributes to EBV-positive BL pathogenesis by inhibiting PRDM1 induction at the onset of plasma cell differentiation in the GC (which is also associated with increased MUM1 expression), analogous to PRDM1 inactivation in the activated B-cell/non-GC B-cell type of DLBCL.3,4 The frequent expression of MUM1/IRF4 in BL is intriguing, and may suggest aberrant expression in early GC BL precursors instead of an indicator of late/post-GC differentiation.
EBV has been shown to induce epigenetic repression of BIM through DNA methylation in the EBV-infected naive B cells, preventing apoptosis.29 However, it is unlikely that EBV directly causes similar methylation in PRDM1, as we did not observe PRDM1 methylation in LCLs. We have considered the possibility that PRDM1 methylation is functional at an early stage of EBV-positive BL pathogenesis and represents only an epigenetic memory in the ultimate tumor cells (in which the repression of PRDM1 is able to be delegated entirely to BCL6). However, if PRDM1 methylation occurs and is indeed important at an earlier stage, we might have expected hypermethylation in higher proportion of tumor cells among primary cases, as the trend during tumorigenesis is generally toward increased methylation, an epigenetic mark that is rather stable. The absence of PRDM1 methylation in LCL cells further argues against it being an early event in BL pathogenesis. Thus, we favor PRDM1 methylation as a relatively late, possibly subclonal event in tumorigenesis of a subset of EBV-positive BLs, possibly after the tumor cells have converted to a latency I program of EBV latency.
How would PRDM1 methylation contribute to pathogenesis or tumor progression in EBV-positive BL? EBV-positive BLs typically have latency I program and express BCL6, a repressor of PRDM1 transcription. We hypothesize that PRDM1 methylation may have functional significance in the scenario when BCL6 in the EBV-positive BL cells is downregulated and signals capable of inducing PRDM1 are present. One of the EBV genes, LMP2A, may be of interest for further investigation because of its potential regulatory functions on both BCL6 and PRDM1 in the latency I setting. Expression of LMP2A is normally associated with EBV latency II and III programs. Interestingly, it can also be detected at low levels by RT-PCR and immunoblotting in endemic BL, whereas LMP1 and EBNAs (except EBNA1) are not detectable.30,31 LMP2A is thought to have a functional role in the development of BL by protecting the cells from MYC-induced apoptosis and promoting early expansion of tumor cells carrying MYC translocation after they exit the GC.32 High levels of LMP2A are subsequently selected against by immune surveillance upon tumor progression, resulting in lower levels of LMP2A expression. Interestingly, LMP2A can activate a variety of pathways, including NF-κB, and increase PRDM1 expression.33 It can also downregulate BCL6 expression through reducing FoXO1 expression.34 Consistent with this, genomic expression profiling of endemic and HIV-related BL demonstrated increased activity of the NF-κB pathway,13 among other signaling pathways in these cells, which has the potential to induce PRDM1 expression.35 LMP2A-mediated PRDM1 induction can potentially be detrimental to the growth of BL cells. Indeed, we have shown in this study that PRDM1 overexpression in BL cell lines resulted in cell cycle arrest. PRDM1 may also lead to terminal differentiation, which can trigger lytic replication in EBV-infected memory B cells.36 Thus, PRDM1 methylation may benefit the survival and maintenance of a subset of EBV-positive BL cells with higher LMP2A expression by limiting PRDM1 expression; hence, this epigenetic alteration is selected for during tumor development. The subclonal detection of PRDM1 methylation in primary EBV-positive BLs may reflect heterogeneity in LMP2A expression. Further experiments are necessary to further confirm this hypothetical model linking LMP2A and PRDM1.
The pathogenesis of EBV-positive BL has been proposed to follow a similar natural history as non-neoplastic EBV-infected B cells, initiating from EBV infection of naive B cells, which adopt a latency III growth program similar to EBV-immortalized LCLs, entry into the GC reaction with acquisition of the critical MYC translocation and the eventual generation of EBV-infected BL cells expressing the same latency program (latency I) as the dividing EBV-infected normal memory B cells.37 PRDM1 appears also to be a target of downregulation in the earlier stages of BL pathogenesis via other mechanisms. Posttranscriptional mechanisms may be responsible for downregulating PRDM1 during the latency III program in EBV-positive B cells. In LCLs, although PRDM1α mRNA is expressed at relatively high levels, PRDM1 protein expression is low (unpublished observations). A recent study demonstrated that the EBV LMP1 protein in infected GC B cells downregulates PRDM1α, thereby inhibiting plasma cell differentiation and entry into the virus lytic cycle and cell death.17 This finding implies that PRDM1 is a virus target for downregulation in EBV-infected B cells in the GC during EBV-positive BL lymphomagenesis. Thus, PRDM1 appears to be a critical target for inhibition during the life cycle of BL development, its functions suppressed by various mechanisms derived by EBV itself or the cellular host.
In summary, our data suggest that inactivation of PRDM1 by hypermethylation of its 5′ regulatory region contributes to the pathogenesis of ~50% of EBV-associated BL. Our findings provide further supportive evidence for the importance of impairment of PRDM1-mediated functions during lymphomagenesis leading to EBV-positive BL. Aggressive natural killer cell malignancies, the vast majority of which are EBV-related, also harbor PRDM1 methylation.10,27 Our study raises the possibility that PRDM1 inactivation has a critical role in EBV-related malignancies in general.
References
Martins G, Calame K . Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol 2008; 26: 133–169.
Tam W . PRDM1 (PR domain containing 1, with ZNF domain). Atlas Genet Cytogenet Oncol Haematol 16: 135–140.
Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 2006; 203: 311–317.
Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM . Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006; 107: 4090–4100.
Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 2010; 18: 580–589.
Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010; 18: 568–579.
Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A et al. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: a potential role of the microRNA let-7. Am J Pathol 2010; 177: 1470–1479.
Shaffer AL, Young RM, Staudt LM . Pathogenesis of human B cell lymphomas. Annu Rev Immunol 2012; 30: 565–610.
Karube K, Nakagawa M, Tsuzuki S, Takeuchi I, Honma K, Nakashima Y et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood 2011; 118: 3195–3204.
Kucuk C, Iqbal J, Hu X, Gaulard P, De Leval L, Srivastava G et al. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc Natl Acad Sci USA 2011; 108: 20119–20124.
Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 2013; 122: 2683–2693.
Swerdlow SH, Campo E, Seto M, Muller-Hermelink HK et al. Mantle cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H (eds) WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, pp 229–232, 2008.
Piccaluga PP, De Falco G, Kustagi M, Gazzola A, Agostinelli C, Tripodo C et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011; 117: 3596–3608.
Bellan C, Lazzi S, Hummel M, Palummo N, de Santi M, Amato T et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood 2005; 106: 1031–1036.
Cattoretti G . PRDM1/BLIMP-1 expression in lymphomas. Haematologica 2006; 91: 434–435.
Leucci E, Onnis A, Cocco M, De Falco G, Imperatore F, Giuseppina A et al. B-cell differentiation in EBV-positive Burkitt lymphoma is impaired at posttranscriptional level by miRNA-altered expression. Int J Cancer 2010; 126: 1316–1326.
Vrzalikova K, Vockerodt M, Leonard S, Bell A, Wei W, Schrader A et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011; 117: 5907–5917.
Li LC, Dahiya R . MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18: 1427–1431.
Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T . BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005; 21: 4067–4068.
Nie K, Gomez M, Landgraf P, Garcia JF, Liu Y, Tan LH et al. MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed–Sternberg cells: a potential pathogenetic lesion in Hodgkin lymphomas. Am J Pathol 2008; 173: 242–252.
Speck SH . EBV framed in Burkitt lymphoma. Nat Med 2002; 8: 1086–1087.
Kluin PM, Harris NL, Stein H, Leoncini L, Raphael M, Campo E et al. B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, pp 265–266, 2008.
Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL . Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol 2004; 173: 1158–1165.
Masucci MG, Contreras-Salazar B, Ragnar E, Falk K, Minarovits J, Ernberg I et al. 5-Azacytidine up regulates the expression of Epstein–Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt's lymphoma line rael. J Virol 1989; 63: 3135–3141.
Robertson KD, Hayward SD, Ling PD, Samid D, Ambinder RF . Transcriptional activation of the Epstein–Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol Cell Biol 1995; 15: 6150–6159.
Hangaishi A, Kurokawa M . Blimp-1 is a tumor suppressor gene in lymphoid malignancies. Int J Hematol 2010; 91: 46–53.
Iqbal J, Kucuk C, Deleeuw RJ, Srivastava G, Tam W, Geng H et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia 2009; 23: 1139–1151.
Cattoretti G, Angelin-Duclos C, Shaknovich R, Zhou H, Wang D, Alobeid B . PRDM1/Blimp-1 is expressed in human B-lymphocytes committed to the plasma cell lineage. J Pathol 2005; 206: 76–86.
Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ . Epstein–Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathogen 2009; 5: e1000492.
Bell AI, Groves K, Kelly GL, Croom-Carter D, Hui E, Chan AT et al. Analysis of Epstein–Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J Gen Virol 2006; 87 (Part 10): 2885–2890.
Tao Q, Robertson KD, Manns A, Hildesheim A, Ambinder RF . Epstein–Barr virus (EBV) in endemic Burkitt's lymphoma: molecular analysis of primary tumor tissue. Blood 1998; 91: 1373–1381.
Bieging KT, Swanson-Mungerson M, Amick AC, Longnecker R . Epstein–Barr virus in Burkitt's lymphoma: a role for latent membrane protein 2A. Cell Cycle 2010; 9: 901–908.
Wang H, Nicholas MW, Conway KL, Sen P, Diz R, Tisch RM et al. EBV latent membrane protein 2A induces autoreactive B cell activation and TLR hypersensitivity. J Immunol 2006; 177: 2793–2802.
Shore AM, White PC, Hui RC, Essafi A, Lam EW, Rowe M et al. Epstein–Barr virus represses the FoxO1 transcription factor through latent membrane protein 1 and latent membrane protein 2A. J Virol 2006; 80: 11191–11199.
Calame K . Activation-dependent induction of Blimp-1. Curr Opin Immunol 2008; 20: 259–264.
Laichalk LL, Thorley-Lawson DA . Terminal differentiation into plasma cells initiates the replicative cycle of Epstein–Barr virus in vivo. J Virol 2005; 79: 1296–1307.
Thorley-Lawson DA, Allday MJ . The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol 2008; 6: 913–924.
Acknowledgements
We acknowledge Sharon Barouk-Fox for her assistance in organization of the materials. This study is supported by a departmental fund from the Department of Pathology and Laboratory Medicine at Weill Cornell to WT.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhang, T., Ma, J., Nie, K. et al. Hypermethylation of the tumor suppressor gene PRDM1/Blimp-1 supports a pathogenetic role in EBV-positive Burkitt lymphoma. Blood Cancer Journal 4, e261 (2014). https://doi.org/10.1038/bcj.2014.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2014.75
This article is cited by
-
Targeting the signaling in Epstein–Barr virus-associated diseases: mechanism, regulation, and clinical study
Signal Transduction and Targeted Therapy (2021)
-
Clinicopathological features of primary thyroid Burkitt’s lymphoma: a systematic review and meta-analysis
Diagnostic Pathology (2020)
-
Differential epigenetic regulation between the alternative promoters, PRDM1α and PRDM1β, of the tumour suppressor gene PRDM1 in human multiple myeloma cells
Scientific Reports (2020)
-
Age-related changes in the BACH2 and PRDM1 genes in lymphocytes from healthy donors and chronic lymphocytic leukemia patients
BMC Cancer (2019)
-
Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia
Journal of Hematology & Oncology (2017)
