
Abstract
Our goal is to develop a rapid and scalable system for functionally evaluating deregulated genes in multiple myeloma (MM). Here, we forcibly expressed human cMYC and KRAS12V in mouse T2 B cells (IgM+B220+CD38+IgD+) using retroviral transduction and transplanted these cells into lethally irradiated recipient mice. Recipients developed plasmacytomas with short onset (70 days) and high penetrance, whereas neither cMYC nor KRAS12V alone induced disease in recipient mice. Tumor cell morphology and cell surface biomarkers (CD138+B220−IgM−GFP+) indicate a plasma cell neoplasm. Gene set enrichment analysis further confirms that the tumor cells have a plasma cell gene expression signature. Plasmacytoma cells infiltrated multiple loci in the bone marrow, spleen and liver; secreted immunoglobulins; and caused glomerular damage. Our findings therefore demonstrate that deregulated expression of cMYC with KRAS12V in T2 B cells rapidly generates a plasma cell disease in mice, suggesting utility of this model both to elucidate molecular pathogenesis and to validate novel targeted therapies.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a B-cell neoplasm characterized by accumulation of monoclonal plasma cells.1 Neoplastic transformation in MM is associated with genomic and epigenetic dysregulation.2 Previous studies have revealed that 40% of MM harbor chromosome translocations, including CCND1, CCND2, cMAF, MAFB and FGFR3/WHSC1, with immunoglobulin heavy chain (IgH).3 Deletions of chromosome 13 are frequently detected in early and late stage MM.4 During disease progression, genetic lesions accumulate, including mutations of NRAS and KRAS, overexpression of cMYC and downregulation of P53.5 Using whole-genome sequencing and whole-exome sequencing,6 more genetic lesions have been identified. Thus, it is critical to develop a short latency in vivo model to functionally evaluate the roles of these dysregulated genes in MM pathogenesis.
Mouse models both facilitate evaluation of the roles of genetic lesions detected in MM and provide for assessing therapeutic agents. The earliest mouse model for MM was induced by intraperitoneal injection of mineral oil, adjuvant and alkanes in BABL/c mice. These mice develop plasmacytomas at 200 days post injection;7 however, plasmacytoma cells typically grow locally at the site of injection and rarely metastasize to bone marrow (BM). Widely used models now include xenograft models of MM generated by subcutaneous injection of human MM cell lines or primary human MM cells into SCID gamma mice. Particularly useful is the SCID-hu model, which is made by directly injecting MM cell lines or patient MM cells into human fetal bone implanted subcutaneously in SCID mice.8 This model provides three-dimensional bone-like scaffolds to mimic the human MM microenvironment and has been used to both assess preclinical drugs and study MM pathogenesis. Another mouse model has been developed by transferring 5T2MM or 5T33MM mouse MM lines into syngeneic recipient mice. These mouse MM lines were established from aged C57BL/KaLwRij mice, which spontaneously develop a plasmacytoma with a low frequency and along with an osteolytic bone disease. These cells can be labeled with bio-trace marker, such as luciferase9 or green fluorescent protein (GFP),10 for in vivo imaging. A similar model was recently developed by intravenous injections of an in vivo-selected MOPC315 cell line into BALB/c mice.11 Several transgenic mouse models have been developed based on expression of cMYC under control of an Ig light chain gene,12 XBP-1,13 cMAF14 or cMYC15 under the control of the Ig VH promoter and enhancer elements. These models recapitulate characteristics of MM; however, they are technically challenging and time consuming with long latency times and do not allow for evaluating multiple gene functions at a time.
A retroviral transduction/transplantation mouse model can overcome these limitations of transgenic mouse models as retroviral vectors can be used to overexpress or silence multiple gene(s) in target cells in a temporal sequence.16 Retroviral transduction/transplantation mouse models have been widely used to study acute myeloid leukemia,17 chronic myeloid leukemia,18 B-cell acute lymphoid leukemia19 and the majority of myeloproliferative neoplasms.20 Based on previous studies,12 we hypothesized that retroviral delivery of cMYC into a later stage B-cell subset might induce plasmacytomas in mice. MYC requires the cooperation or complementation with other oncogenes, such as v-H-ras or v-raf, for transformation.21 We here therefore introduced KRAS (KRAS12V) mutation to complement the function of cMYC in tumorigenesis. With this strategy, we generated a rapid-onset high-penetrance plasmacytoma mouse model by enforced expression of cMYC and KRAS12V in later transition (T2) B-cell subset. This model provides a rapid tool to functionally evaluate genes in MM pathogenesis, as well as evaluate novel targeted therapies.
Materials and methods
DNA constructs
All PCR products were cloned into T-easy vector (Promega BioSystems, Sunnyvale, CA, USA) and completely sequenced to verify the correct reading frame.
To make a subclone vector MSCVm, MSCV-IRES-eGFP (MIG) was digested with BglII and CalI to remove IRES and eGFP, and ligased with an oligonucleotide (5′-GATCCGAATTCGTTAACGGATCCGGGCCCAAGCTTCAATTGGCGGCCGCAGATCTCTCGAGAT-3′) containing multiple clone sites, EcoRI, HpaI, BamHI, ApaI, HindII, MfeI, NotI, BglII, XhoI and ClaI.
Vector MIGm was constructed by introducing IRES-eGFP generated by PCR with a 5′ primer containing an EcroR1 and a 3′ primer containing a MfeI site into the MfeI site of MSCVm vector. MIGm-cMyc was generated by introducing cMYC released from MSCV-hcMYC-IRES-GFP vector (Addgene, Cambridge, MA, USA) with EcoRI into EcoRI site of MIGm vector. MIGm-KRAS12V was made by inserting KRAS12V obtained from T vector with NotI (blunt) and MfeI into MIGm vector disgested with EcoR1 and HpaI.
MSCVm-cMYC-2A-eGFP-IRES-KRAS12V (MIKMG) was made by the following process: first, MSCVm was inserted a 2A sequence (5′-GATCCCAGCTGTTGAATTTTGACCTTCTTAAGCTTGCGGGAGACGTCGAGTCCAACCCCGGGCC-3′), which contains a BamHI and an ApaI competent site at 5′ and 3′ end, respectively; then, an eGFP coding sequence was inserted into MSCVm-2A vector, which was generated by PCR with a 5′ primer containing a ApaI site without its start code sequence and 3′ primer containing an MfeI site; next, IRES element was cloned into MSCVm-2A-eGFP vector at MfeI site, which was generated by PCR with a 5′ primer containing an EcoR1 site and 3′ primer containing an MfeI site; the open reading frame encoding cMYC generated by PCR from MIG-hcMYC vector (Addgene, 18119), with a 5′ primer containing an EcoR1 site and a 3′ primer containing a BamH1 site without stop coding sequence, was inserted into MSCVm-2A-eGFP-IRES with EcoRI and BamHI. KRAS12V open reading frame generated by PCR from pBabe-KRAS12V (Addgene, 12544) was cloned into MSCVm-cMYC-2A-eGFP-IRES with MfeI and NotI behind the IRES element.
Viral stock
All DNAs were purified in CsCl gradients. Lenti-X 293T cells (Clontech Laboratories, Mountain View, CA, USA) were co-transfected with 10 μg retroviral vector DNA and 5 μg pCL-Ecotropic packaging vector DNA using CaCl2 method. Medium was changed at 24 h post transfection, and the supernatant was harvested at 48 h after transfection. Supernatant was filtered with 0.45-μm filters, aliquoted and frozen in a freezer at −80 °C. The virus titer was determined by transduction of NIH 3T3 cells, and the percentage of GFP+ cells was screened by flow cytometry. After calculation, all viruses had titers >4.8 × 106 GFP+/ml.
Mouse IgM+ B-cell isolation
All animal experiments were approved by and conformed to the standards of the Institutional Animal Care and Use Committee at the DFCI. BALB/c or C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME, USA; 000651) aged 6–12 weeks were used in all experiments. Mice were killed by CO2 asphyxiation. Mouse IgM+ splenic B cells were isolated using magnetic microbeads and LS MACS separation columns (Miltenyi Biotec, Cambridge, MA, USA).
Retroviral transfection and transplantation
IgM+ cells were counted and plated at 2 × 107 cells per 10-cm plate in pre-stimulation medium of RPMI1640 containing 15% (vol/vol) inactivated fetal calf serum, 1% (vol/vol) penicillin/ streptomycin, 1.0 μg/ml ciprofloxacin, 200 μM l-glutamine, 10 ng/ml recombinant murine interleukin 4 (mIL4; Sigma, St Louis, MO, USA), and 50 μg/ml lipopolysaccharide (LPS; Sigma). After pre-stimulation for 24 h, viable cells were counted and transduced with retroviral stocks in the same medium containing 50% retroviral supernatant, 10 mM Hepes, pH 7.4 and 2 μg/ml polybrene. To increase transduction efficiency, virus and cells were cosedimented at 1000 g for 90 min in a Sorvall RT-5.3 centrifuge. Medium was changed after a 3-h adsorption period. On the next day, a second round of transduction and cosedimentation was performed using the same conditions. After another 3 h, cells were harvested and 5 × 105 cells were injected via the lateral tail vein with 5 × 105 BM into lethally irradiated recipients.
In vitro proliferation and differentiation assays
For colony assay, 1 × 105 transfected cells in 100 μl were mixed with 3 ml HSC-CUF basic media (Miltenyi Biotec, 130-091-275) in 15-ml tube. Cells were transferred into six-well cell culture plates with 16-gauge blunt-end needles (Miltenyi Biotec, 130-091-558), and the plate’s interspace was filled with 10 ml sterilized phosphate-buffered saline (PBS) to maintain humidity of the culture environment. Cells were cultured at 37 °C and 5% CO2 for 2 weeks.
Histopathology and immunohistochemistry (IHC)
Tissues were fixed, processed, sectioned and stained with hematoxylin-eosin by routine methods. Femurs were additionally treated for 1 h in decalcifying solution (Fisher Scientific, Cambridge, MA, USA). IHC was performed according to the standard procedures using rat anti-mouse CD45R/B220 (RA3-6B2) and rat anti-mouse CD138 (281-2) monoclonal antibodies obtained from BD Biosciences; anti-cMYC, RAS and eGFP antibodies obtained from Cell Signaling (Franklin Lakes, NJ, USA); as well as goat anti-mouse polyclonal antibodies for IgH chains IgA, IgM, IgG and kappa or lambda light chains conjugated to horseradish peroxidase (Southern Biotechnology, Birmingham, AL, USA).
Flow cytometry
Red blood cells were removed from single-cell suspensions of peripheral blood, BM, peritoneal fluid and spleen by RBC lysis buffer. Cells were then washed once with PBS and stained for 15 min at room temperature with the combination of the following antibodies: IgM-PE, B220-PECy7, CD138-APC, CD38-PE and IgD-PE washing once with PBS, and then cells were analyzed on a FACScalibur machine (Becton Dickinson, Franklin Lakes, NJ, USA) using Cell Quest software (Becton Dickinson).
Western blots
Whole-cell extracts from target cells were prepared, electroblotted onto NC membranes (Amersham, Wilson, OK, USA) and probed with primary antibodies according to the standard procedures. The anti-cMYC, RAS and eGFP antibodies were obtained from Cell Signaling. Following incubation with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibody (Cell Signaling, Danvers, MA, USA), bound Igs were detected using ECL detection solutions (Pierce, Rockford, IL, USA). Anti-GAPDH served as a loading control.
Southern blot analysis of the IgH gene
Genomic DNA was prepared from IgM+ B cells or cMYC/KRAS12V-induced plasmacytoma cells with Qiagen DNeasy 96 Blood and Tissue Kit (Qiagen, Valencia, CA, USA). DNA was digested with EcoRI. Blots were hybridized with a JH4 probe of the mouse IgH locus. The JH4 probe was generated by PCR using a plasmid containing a 1.9 BamHI-EcoRI genomic fragment of mouse heavy-chain locus as template and primers JH4f (5′-TACTATGCTATGGACTACTGG-3′) and JH4r (5′-CTCTCCAGTTTCGGCTGAATC-3′). Southern blot hybridization was performed as described.22
Gene expression profiling
RNA from purified IgM+ B cells and plasmacytoma cells was extracted with RNA mini kit (Grand Island, NY, USA), and gene expression profiling was performed using the Affymetrix mouse 430A2.0 gene chip (Affymetrix, Cleveland, OH, USA). Microarray data were analyzed with OneChannelGUI package in R workplace. Differential expression was determined using the LIMMA model. Gene signatures were analyzed with gene set enrichment analysis (GSEA) in Molecular Signatures Database (MSigDB).
Results
Design of multiple gene expression MSCV-based vectors
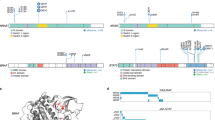
To develop an adoptive mouse plasmacytoma model, we purified splenic IgM+ B cells, which were cultured with LPS and mIL4 for 24 h. These target cells were transfected twice within 24 h and then injected via tail vein (intravenous) into lethally irradiated syngeneic recipients (Figure 1a). To develop oncogene expression vectors, we modified the retroviral vector MIG (MSCV-IRES-eGFP) by inserting multiple cloning sites. We constructed three oncogene expression vectors: cMYC (5′LTR-cMYC-IRES-eGFP-3′LTR), KRAS12V (5′LTR-KRAS12V-IRES-eGFP-3′LTR) and cMYC/KRAS12V (5′LTR-cMYC-2a-eGFP-IRES-KRAS12V-3′LTR). All oncogenes are driven by the cytomegalovirus type I enhancer and the mouse sarcoma virus promoter within the 5′ LTR (long terminal repeat; Figure 1b). Target genes were efficiently expressed by the vectors in transiently transfected 293T cells (Figure 1c).

cMYC/KRAS12V can transform BaF3 cells independent of IL3 and transduce T2 B cells in vitro. (a) Workflow for generation of the adoptive plasmacytoma mouse model. (b) Schematic diagram of MSCV-based retroviral vectors: MIG, cMYC, KRAS12V and cMYC/KRAS12V. (c) Western blot analysis showed expression of MIG, cMYC, KRAS12V and cMYC/KRAS12V in transfected 293T cells. Total protein was analyzed against human MYC (upper) and RAS (middle); GAPDH (lower) served as a loading control. (d) KRAS12V and cMYC/KRAS12V drove BaF3 cells to grow independent of IL3 in vitro. Representative cells from two independent experiments are shown. (e) The purification of mouse IgM+ spleen B cells after micro-bead isolation is shown (left panel). The cell population transited to T2 B-cell subset (IgM+B220+ CD38+IgD+) after stimulation with LPS and mIL4 for 48 h (right three panels). Assays were independently and repeatedly performed. (f) Colonies in methylcellulose assays were observed only in cells transduced by cMYC/KRAS12V but not other groups. Three independent experiments were performed.
To investigate the biological and functional sequelae of these oncogenes, we transfected BaF3 cells with MIG, cMYC, KRAS12V or cMYC/KRAS12V. Two days after transfection, GFP+ cells were sorted and cultured without IL3. After a week, both KRAS12V and cMYC/KRAS12V, but not cMYC or MIG alone, drove BaF3 cells growth in vitro independent of IL3 (Figure 1d). These data suggest that KRAS12V has the capacity to suppress cMYC-induced apoptosis in BaF3 cells.
Previous studies have shown that overexpression of oncogenes cMYC, cMAF and XBP-1 driven by Eμ promoter in immature B cells induces plasma cell neoplasms in mice.12, 13, 14, 15 Here we used T2 B-cell subset (IgM+B220+CD38+IgD+)23 as target cells, which are generated from stimulating purified IgM+ B cells with LPS and mIL4 for 48 h (Figure 1e). To determine whether cMYC-, KRAS12V- or cMY/KRAS12V-transfected cells could grow in vitro, we performed soft agar colony assays. T2 B cells transfected with MIG, cMYC, KRAS12V or cMYC/KRAS12V were seeded in soft agar culture media, and after 2 weeks, only cMYC/KRAS12V-transduced cells formed colonies in vitro (Figure 1f). These results suggested that cMYC/KRAS12V had the capacity to promote independent T2 B cells growth in vitro.
cMYC/KRAS12V induced plasmacytomas in BALB/c mice
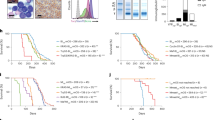
To determine whether cMYC/KRAS12V can transform T2 B cells and induce plasmacytomas in vivo, we transplanted T2 B cells transfected with MIG, cMYC, KRAS12V or cMYC/KRAS12V into lethally irradiated syngeneic recipient mice. Only mice that received cMYC/KRAS12V-transfected T2 B cells developed fatal tumors within 10 weeks post transplantation (Figure 2a). In diseased mice, peritoneal tumor, splenomegaly, and ascites were noted (Figure 2b). Tumor cells had dispersed nuclear chromatin, a low nuclear-to-cytoplasmic ratio and amphophilic cytoplasm with paranuclear hof. Binucleate cells and mitoses were rarely observed (Figure 2c). The analyses by flow cytometry showed tumor cell surface positivity for GFP and CD138, but absence of IgM and B220 (CD138+B220−IgM−GFP+) (Figure 2d). These features and phenotypes resemble previously reported transgenic and chemically induced plasmacytoma mouse models.24, 25 We also tested whether cMYC/KRAS12V-transformed plasmacytoma cells could cause similar tumors in secondary transplant recipients. In these experiments, splenic tumor cells from diseased mice were transferred into syngeneic mice, and all recipients developed tumors within 4 weeks (Figure 2a).

cMYC/KRAS12V-induced plasmacytoma in BALB/c mice. (a) Recipients receiving MIG- or cMYC- or KRAS12V-transfected donor cells remained tumor free, whereas all recipients of cMYC/KRAS12V cells died with plasmacytomas. Secondary transplantation recipients of cMYC/KRAS12V-transduced tumor cells died with similar syndromes (2nd, secondary transplantation). Primary transplantation was repeated five times, and secondary transplantation was independently and repeatedly performed. Group sizes and survival times are indicated. (b) Tumor in peritoneal cavity and splenomegaly were observed in the cMYC/KRAS12V group animals (n=20, from five independent experiments). (c) H&E staining showing tumor cell morphology (n=6). (d) Tumor cells from peritoneal cavity (P.C.) and ascites (A.C.) (n=10) were characterized as CD138+B220−IgM−GFP+ by flow cytometry.
In chemically induced plasmacytoma mouse models, plasmacytoma cells are typically located at the site of injection and infrequently metastasize to BM and other organs.26, 27 In MM patients, tumor cells frequently affect BM and kidney, but rarely directly infiltrate other organs. To further define the anatomic distribution of plasmacytoma cells in our model, cells isolated from the peripheral blood, BM and spleen were analyzed for eGFP and CD138 expression. We found that plasmacytoma cells infiltrated BM (30–50%) and spleen (10–70%) and, to a lesser (⩽ 0.3% of total cells) extent, in peripheral blood (Figure 3a). Only cMYC/KRAS12V mice developed plasmacytomas with tumor cell infiltration in the BM and spleen. All tumor cells have similar cell morphology and surface phenotype (Figures 3b and c).

Plasmacytoma cells infiltrated multiple organs. (a) Flow cytometry analysis of GFP and CD138 to track tumor cells in peripheral blood (PB), femur and tibia BM and spleen (SPL). Numbers represent tumor cell percentage in respective gates. Samples (n=25) of individual mice from different experiments were analyzed. (b) BM and spleen (SPL) sections from mice receiving MIG-, cMYC-, KRAS12V- and cMYC/KRAS12V-transfected cells were stained with hematoxylin-eosin (H&E) and anti-CD138 antibody to identify plasmacytoma cells. (c) Liver (LV) sections from diseased mice were stained with H&E (left) and anti-CD138 antibody (right). (d) IHC analysis of BM sections showed expression of cMYC, KRAS and eGFP in cMYC/KRAS12V-induced plasmacytoma cells.
To determine the contribution of cMYC and KRAS12V in plasmacytoma development, we performed IHC assays with antibodies specific for cMYC, KRAS and GFP on BM specimens. Positive staining for cMYC, KRAS and GFP were confirmed in plasmacytoma cells within tissues from cMYC/KRAS12V, but not MIG, cMYC or KRAS12V, mice (Figure 3d).
Hypergammaglobulinemia, renal and bone alterations in diseased mice
A characteristic feature of MM and other plasma cell neoplasms is secretion of monoclonal Ig, detected as a distinct band (M-spike) by serum protein electrophoresis. In our model, M-spikes were detected in both the serum and ascites in cMYC/KRAS12V mice, but not in the serum of others (MIG, cMYC and KRAS12V) (Figure 4a and data not shown). The Ig chain isoforms in diseased mice include IgG1, IgM and IgA heavy chains and both of κ and λ light chains (Figure 4b). Southern blot analysis of IgH gene rearrangement in plasmacytoma cells indicated that cMYC/KRAS12V-induced plasmacytomas were clonal (Figure 4c).

Evidence of clonality and hypermutation of expressed Ig genes in cMYC/KRAS12V-induced plasmacytoma mice. (a) Serum protein electrophoresis (SPEP) in serum and ascites from cMYC/KRAS12V-induced plasmacytoma mice. The bands are indicated with arrows. Samples were obtained from all the group animals as labeled (n=5 each group). (b) Ig isoform identification. Ascites were collected from plasmacytoma mice and analyzed with Thermo Scientific Pierce Rapid ELISA Mouse mAb Isotyping Kit. The Ig isoforms (n=10) are represented by color labeled bands as indicated. (c) Southern blot analysis for clonotypic IgH -chain rearrangement in plasmacytoma cells. Genomic DNA was isolated from IgM+ B cells (sample 1) or cMYC/KRAS12V-induced plasmacytoma cells (samples 2–7), digested with EcoRI restriction enzyme and hybridized with murine JH4 probe. GL denotes germline band.
In human MM, development of Ig heavy and light chain, as well as infiltration of tumor cells, leads to both renal tubular obstruction and glomerular damage. We observed glomerular shrinkage in cMYC/KRAS12V mice but not in other groups (Figure 5a). These renal lesions were similar to pathological manifestations in MM.28 To assess Ig deposition, we performed IHC assays with antibodies specific for mouse Igs IgG, IgM, κ and λ. Ig chains were deposited in both the tubules and glomerulus (Figure 5b).

Renal and bone alterations in cMYC/KRAS12V-induced plasmacytoma mice. (a) Renal tissue from mice receiving MIG-, cMYC-, KRAS12V- and cMYC/KRAS12V-transfected cells (n=3 each group) were analyzed by light microscopy (hematoxylin-eosin staining) and IHC staining with antibodies against mouse Ig κ chains. (b) Histological sections of kidneys of mice receiving cMYC/KRAS12V-transduced plasmacytomas were analyzed by IHC staining with antibodies against mouse Ig κ and λ light chains, as well as IgG and IgM heavy chains. (c) Micro-CT analysis demonstrated no bone lesions in all the group animals. Representative graphs from three individual animals are shown.
Another hallmark of human MM is osteolytic bone lesions. To determine whether the bone lesions developed in diseased mice, we examined bone structure changes with micro-computed tomography (micro-CT). No bone osteolysis was detected, even in the cMYC/KRAS12V group mice (Figure 5c).
Gene expression profiling in cMYC/KRAS12V-induced plasmacytoma cells
To gain further insights into the roles of cMYC/KRAS12V in tumorigenesis and identify the tumor type, we analyzed the gene expression signatures of cMYC/KRAS12V-induced tumor cells by using global transcriptional profiling analysis and comparing tumor cells to syngeneic murine IgM+ B cells. Genes were considered significantly altered based on more than twofold change in mean expression (P<0.05). Using these criteria, 4466 genes were significantly altered in tumor cells: 2333 were upregulated and 2133 were downregulated. Transcription factors required for plasma cell differentiation and survival were significantly increased: PRDM1 (Blimp-1), IRF4 and XBP-1 were increased 5.9-, 4.8- and 3.6-fold, respectively. Conversely, B-cell transcription factors absent or downregulated in plasma cells, including BCL-6, CIITA, MTA3, EBF1, STAT3, PAX5 and SPIB, were also significantly downregulated in plasmacytoma cells (Supplementary Data, Supplementary Table S1). Expression of several hallmark plasma cell differentiation and survival genes were confirmed using quantitative real-time PCR (Figure 6a). GSEA also revealed significant enrichments for upregulation of genes related to plasma cell differentiation and survival (Figure 6b, Supplementary Figure S1A), whereas gene sets related to B-cell receptor signaling were downregulated (Figure 6d). As expected, genes regulated by IRF4 and XBP1 were significantly upregulated (Figure 6e and Supplementary Figures S1B and C). Finally, several components of the nuclear factor-κB (NF-κB) pathways, including NF-κB1 and NF-κB2, and Rel, RelA and RelB were also downregulated in tumor cells (Figure 6f and Supplementary Figure S1D). These data are consistent with previous comparison of plasma cells versus IgM+ B cells.29, 30, 31, 32

Plasma cell transcription signatures in cMYC/KRAS12V-transduced plasmacytoma cells. (a) Expression of selected genes from microarray results were confirmed using quantitative real-time PCR. All data represent mean of triplicate experiments. (b) GSEA showed a plasma cell gene expression signature of plasmacytoma cells. (c) GSEA showed genes associated with B-cell receptor signaling to be significantly downregulated in plasmacytoma cells. (d) GSEA showed genes in IRF4 target gene set were upregulated in plasmacytoma cells. (e) Table of the gene sets related to plasma cell differentiation and survival.
To confirm the functional roles of cMYC and KRAS12V in plasmacytoma genesis, we performed GSEA with four well-established canonical transcriptional signatures of MYC-dependent genes.26, 33, 34, 35 Most genes in these gene sets were significantly upregulated in plasmacytoma cells (Figure 7a and Supplementary Figure S2A). Previous studies have demonstrated that RAS suppressed cMYC-induced apoptosis via activating PI3K/AKT pathway.36 Activated AKT in turn phosphorylates and activates mammalian target of rapamycin (mTOR), an essential component of mTORC1 that promotes tumor cell proliferation and survival.37 In cMYC/KRAS12V-transduced plasmacytoma cells, genes in mTOR target gene set38 were significantly upregulated (Figure 7b and Supplementary Figure S2B). These results further confirm the formation of plasmacytoma due to cooperation of cMYC and KRAS12V. As in other tumors, genes regulating cell cycle,39 as well E2F40 target genes, and genes suppressed by RB141 were also significantly upregulated (Supplementary Figure S2C). Apoptotic signaling was inhibited by cMYC/KRAS12V: specifically, genes suppressed by P53 and P73 during cell growth arrest and apoptosis were enriched and upregulated in plasmacytoma cells (Figure 7c and Supplementary Figure S2C). Several other gene sets were also enriched and upregulated plasmacytoma cells, including EZH2,42 NRF243 and YAP44 (Figure 7d). These results provide further insight into the role of cMYC/KRAS12V in plasmacytoma pathogenesis and identify potential therapeutic targets.

Gene expression signatures of cMYC/KRAS12V-transduced plasmacytoma cells. (a) Using GSEA, four gene sets associated with MYC activation are presented in plasmacytoma cells compared with IgM+ B cells. (b) GSEA showed genes upregulated by mTOR in plasmacytoma cells. (c) GSEA showed that genes regulating cell cycle are significantly enriched in plasmacytoma cells. (d) Table of gene sets of MYC, mTOR, cell cycle and cancer-related pathways (from top to bottom) in cMYC/KRAS12V-transduced plasmacytoma cells.
Identifying provirus integration sites and altered genes
Single oncogene transgenic MM mouse models may have long disease latency until the development of disease, as additional genetic lesions and/or epigenetic dysregulations in tumor-initiating cells may be required for tumor development.12, 13 In an adoptive model, provirus integration into host genome can alter host-cell gene expression.45 To determine whether provirus integrations are involved in the pathogenesis in our model, we identified MSCV integration sites using ligation-mediated PCR method. In total, we identified 11 retroviral integration sites in three diseased mice (n=3, 2–4 clones in each tumor sample Figure 8a). The integration sites are not recurrent, and there is no evidence indicating an association with MM (Figure 8b). These data indicate that the plasmacytomas derived by MSCV-cMYC/KRAS12V did not require additional genetic lesions.

Provirus integration sites. (a)The splinkerette-PCR results of provirus integration sites. The lanes of 3% agarose gel showed the splinkerette-PCR production for three mouse plasmacytoma samples with 100 bp marker. Each band represents a provirus integration site and a tumor cell subclone, except for the top band, which represents retroviral vector endogenous fragment (617 bp, not including the linker sequence). (b) Identification of MSCV integration sites in plasmacytoma cells.
Discussion
Here we report a rapid-onset high-penetrance mouse model of plasmacytoma based on enforced expression of both cMYC and KRAS12V in T2 B subset population cells via retroviral transduction, followed by transplantation into lethally irradiated mice. The disease latency was about 7 weeks, all recipients developed disease, and most recipients died within 10 weeks. Compared with transgenic models, this adoptive model significantly reduces both time and cost and provides a highly efficient system to evaluate oncogenes. Moreover, it is feasible to combine oncogenes to study their cooperation effects. Conversely, we can introduce RNA-interfering techniques to suppress the expression of specific targets associated with oncogenes. This rapid model therefore provides a faithful in vivo system to functionally evaluate the genetic lesions in cancer, both elucidating pathogenesis and evaluating therapeutic targets.
We selected cMYC and KRAS12V as examples to examine the possibility that enforced expression of oncogenes in a specific B-cell population could model plasmacytoma in mice. Deregulated activity of cMYC is highly associated with MM,26 and previous transgenic mouse models and chemically induced plasmacytomas in BALB/c mice have demonstrated the roles of cMYC in the development of plasmacytomas.46 KRAS as a secondary oncogene was selected as KRAS mutations can suppress cMYC-induced apoptosis in rat fibroblasts via PI3K/AKT signaling, and KRAS mutations are frequently detected in MM.36 Moreover, previous studies showed that KRAS12V could enhance cMYC-induced apoptosis via constitutively activating Raf pathway in rat fibroblasts.36 Of note, v-myc and v-raf had synergistic roles in plasmacytomas in BALB/c mice induced by pristine priming,21, 47 suggesting that the role of Raf signaling might vary with cell types or genetic backgrounds. As expected, KRAS12V efficiently suppressed cMYC-induced apoptosis in a pre-B-cell line (murine BaF3 cells) in vitro and successfully induced plasmacytomas in vivo by cooperating with cMYC. Finally, in our adoptive mouse model, neither cMYC nor KRAS12V alone was sufficient to induce plasmacytomas in BALB/c mice. Previous transgenic mouse models already indicate that secondary mutations are required for the development of plasmacytomas contributing to their long latency time.13 Here, we provide KRAS12V as a required secondary gene lesion, which significantly reduces disease latency time.
The target cell population is critical for successful induction of plasmacytoma in recipients. Purified splenic IgM+ population includes immature B cells, early transitional B cells (T1 B cells), later transitional B cells and memory B cells.23 After stimulation with LPS and mIL4, the major cell population transits to T2 B-cell subset (IgM+B220+CD38+IgD). In this model, T2 B cells were transformed by cMYC/KRAS12V and induced plasmacytoma in vivo based on both cell morphology and cell surface biomarkers (CD138+B220−IgM−GFP+). Gene profiling also indicated that tumor cells acquire plasma cell and lose IgM+ B-cell gene expression signatures. Specifically, plasma cell differentiation genes were significantly upregulated, whereas B-cell-specific transcription factors were repressed. These results are consistent with previous gene profiling studies comparing plasma cells with IgM+ B cells.29
Although the adoptive mouse model does not accurately reflect all the features of human MM, it recapitulates many major characteristics of the disease. In our cMYC/KRAS12V-induced plasmacytoma mouse model, some mice developed hind limb paralysis with plasma cell infiltration of the spine. Histological examination of femur also showed tumor cell infiltration at multiple loci in BM. The lack of lytic bone lesions observed with micro-CT may be associated with lethal irradiation48 or could be manifested at later points of time. Other MM features, such as hyperglobulinemia and renal injury, were mimicked in this model.
After secondary transplantation of cMYC/KRAS12V-induced plasmacytomas, all recipients developed disease at short latency, 21 days post transplantation compared with 70 days primary transplantation recipients. The major disease features of secondary transplant recipients resemble those in primary transplant recipients. Thus, secondary transplant recipients may represent an even more suitable system for preclinical drug evaluations.
Previous studies have demonstrated that chemically induced plasmacytoma are mouse strain specific.49 Consistent with these findings, we find that BALB/c, but not C57BL/6, mice are susceptible to cMYC/KRAS12V-induced plasmacytoma. This provides us a way to investigate the role of tumor-suppressor genes in MM pathogenesis, such as P53 and RB1. For example, in both human MM and murine plasmacytoma, loss of function of P53 or RB1 is commonly observed. Thus, our model could be used to determine whether P53 or RB1 has a critical role in preventing the development of cMYC/KRAS12V-induced plasmacytoma.
In summary, this adoptive plasmacytoma mouse model faithfully reflects most of the major characteristics of MM in patients. It affords a system to evaluate the oncogenic events in MM, elucidate mechanism of these events and identify potential targets for future therapy. It will also be useful to study microenvironmental and epigenetic factors in MM, as well as assess potential targeted therapies.
References
Kyle RA, Rajkumar SV . Multiple myeloma. N Engl J Med 2004; 351: 1860–1873.
Kuehl WM, Bergsagel PL . Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2002; 2: 175–187.
Bergsagel PL, Kuehl WM . Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol 2005; 23: 6333–6338.
Shaughnessy J, Tian E, Sawyer J, Bumm K, Landes R, Badros A et al. High incidence of chromosome 13 deletion in multiple myeloma detected by multiprobe interphase FISH. Blood 2000; 96: 1505–1511.
Bergsagel PL, Kuehl WM . Chromosome translocations in multiple myeloma. Oncogene 2001; 20: 5611–5622.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467–472.
Anderson PN, Potter M . Induction of plasma cell tumours in BALB-c mice with 2,6,10,14-tetramethylpentadecane (pristane). Nature 1969; 222: 994–995.
Urashima M, Chen BP, Chen S, Pinkus GS, Bronson RT, Dedera DA et al. The development of a model for the homing of multiple myeloma cells to human bone marrow. Blood 1997; 90: 754–765.
Mori Y, Shimizu N, Dallas M, Niewolna M, Story B, Williams PJ et al. Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004; 104: 2149–2154.
Oyajobi BO, Munoz S, Kakonen R, Williams PJ, Gupta A, Wideman CL et al. Detection of myeloma in skeleton of mice by whole-body optical fluorescence imaging. Mol Cancer Ther 2007; 6: 1701–1708.
Hofgaard PO, Jodal HC, Bommert K, Huard B, Caers J, Carlsen H et al. A novel mouse model for multiple myeloma (MOPC315.BM) that allows noninvasive spatiotemporal detection of osteolytic disease. PLoS One 2012; 7: e51892.
Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008; 13: 167–180.
Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007; 11: 349–360.
Morito N, Yoh K, Maeda A, Nakano T, Fujita A, Kusakabe M et al. A novel transgenic mouse model of the human multiple myeloma chromosomal translocation t(14;16)(q32;q23). Cancer Res 2011; 71: 339–348.
Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, Polakiewicz RD et al. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J Clin Invest 2004; 113: 1763–1773.
Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 2011; 145: 145–158.
Cuenco GM, Nucifora G, Ren R . Human AML1/MDS1/EVI1 fusion protein induces an acute myelogenous leukemia (AML) in mice: a model for human AML. Proc Natl Acad Sci USA 2000; 97: 1760–1765.
Daley GQ, Van Etten RA, Baltimore D . Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990; 247: 824–830.
Yu D, Thomas-Tikhonenko A . A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene 2002; 21: 1922–1927.
Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One 2006; 1: e18.
Alexander WS, Adams JM, Cory S . Oncogene cooperation in lymphocyte transformation: malignant conversion of E mu-myc transgenic pre-B cells in vitro is enhanced by v-H-ras or v-raf but not v-abl. Mol Cell Biol 1989; 9: 67–73.
Xie P, Stunz LL, Larison KD, Yang B, Bishop GA . Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 2007; 27: 253–267.
Sanz I, Wei C, Lee FE, Anolik J . Phenotypic and functional heterogeneity of human memory B cells. Semin Immunol 2008; 20: 67–82.
Zhang SL, DuBois W, Ramsay ES, Bliskovski V, Morse HC 3rd, Taddesse-Heath L et al. Efficiency alleles of the Pctr1 modifier locus for plasmacytoma susceptibility. Mol Cell Biol 2001; 21: 310–318.
Silva S, Kovalchuk AL, Kim JS, Klein G, Janz S . BCL2 accelerates inflammation-induced BALB/c plasmacytomas and promotes novel tumors with coexisting T(12;15) and T(6;15) translocations. Cancer Res 2003; 63: 8656–8663.
Kobayashi H, Potter M, Dunn TB . Bone lesions produced by transplanted plasma-cell tumors in BALB/c mice. J Natl Cancer Inst 1962; 28: 649–677.
Potter M, Fahey JL, Pilgrim HI . Abnormal serum protein and bone destruction in transmissible mouse plasma cell neoplasm (multiple myeloma). Exp Biol Med 1957; 94: 327–333.
Korbet SM, Schwartz MM . Multiple myeloma. J Am Soc Nephrol 2006; 17: 2533–2545.
Underhill GH, George D, Bremer EG, Kansas GS . Gene expression profiling reveals a highly specialized genetic program of plasma cells. Blood 2003; 101: 4013–4021.
Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002; 17: 51–62.
Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K . BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat Immunol 2000; 1: 526–532.
Lin Y, Wong K, Calame K . Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 1997; 276: 596–599.
Schlosser I, Holzel M, Hoffmann R, Burtscher H, Kohlhuber F, Schuhmacher M et al. Dissection of transcriptional programmes in response to serum and c-Myc in a human B-cell line. Oncogene 2005; 24: 520–524.
Schuhmacher M, Kohlhuber F, Holzel M, Kaiser C, Burtscher H, Jarsch M et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res 2001; 29: 397–406.
Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV . An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 2003; 4: R69.
Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997; 385: 544–548.
Zoncu R, Efeyan A, Sabatini DM . mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12: 21–35.
Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 2004; 10: 594–601.
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2004; 2: E7.
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 2002; 16: 245–256.
Eguchi T, Takaki T, Itadani H, Kotani H . RB silencing compromises the DNA damage-induced G2/M checkpoint and causes deregulated expression of the ECT2 oncogene. Oncogene 2007; 26: 509–520.
Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K . Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006; 20: 1123–1136.
Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 2010; 38: 5718–5734.
Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147: 759–772.
Coffin JMHS, Varmus HE . Retrovirus. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997.
Potter M, Wiener F . Plasmacytomagenesis in mice: model of neoplastic development dependent upon chromosomal translocations. Carcinogenesis 1992; 13: 1681–1697.
Kurie JM, Morse HC 3rd, Principato MA, Wax JS, Troppmair J, Rapp UR et al. v-myc and v-raf act synergistically to induce B-cell tumors in pristane-primed adult BALBC mice. Oncogene 1990; 5: 577–582.
Seed TM, Chubb GT, Tolle DV . Sequential changes in bone marrow architecture during continuous low dose gamma irradiation. Scanning Microsc 1981; 4: 61–72.
Zhang S, Ramsay ES, Mock BA . Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16INK4a and p19ARF, is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc Natl Acad Sci USA 1998; 95: 2429–2434.
Acknowledgements
We thank Weihua Song, Teru Hideshima and Yu-Tzu Tai for scientific discussion; as well as the Microarray Core and the Flow Cytometry Core Facilities at Dana-Farber Cancer Institute for outstanding technical support. This study was supported in part by National Institutes of Health Grants P50-100707, P01-78378 and RO1-50947. KCA is an American Cancer Society Clinical Research Professor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Blood Cancer Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hu, Y., Zheng, M., Gali, R. et al. A novel rapid-onset high-penetrance plasmacytoma mouse model driven by deregulation of cMYC cooperating with KRAS12V in BALB/c mice. Blood Cancer Journal 3, e156 (2013). https://doi.org/10.1038/bcj.2013.53
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2013.53
Keywords
This article is cited by
-
Adoptive B-cell transfer mouse model of human myeloma
Leukemia (2016)
-
Transgenic mouse model of IgM+ lymphoproliferative disease mimicking Waldenström macroglobulinemia
Blood Cancer Journal (2016)
-
FOXM1 is a therapeutic target for high-risk multiple myeloma
Leukemia (2016)
-
CSNK1α1 mediates malignant plasma cell survival
Leukemia (2015)

{kind=link}
{kind=link}