
Abstract
In this study, we have identified the growth factors supporting myeloma self-renewal in eight myeloma cell lines. All cell lines able to form self-colonies displayed constitutive P-AKT and P-ERK1,2 but not P-STAT3 and did not express CD45, suggesting the presence of an insulin-like growth factor 1 (IGF1) loop. We showed that a blocking anti-insulin-like growth factor 1 receptor (IGF1R) monoclonal antibody (mAb) inhibited colony formation in correlation with IGF1R expression and decreased P-AKT. Imatinib or a blocking anti-stem cell factor (SCF) mAb also inhibited colony formation of two cell lines expressing C-KIT and SCF, and decreased P-AKT. Moreover, the PI3K/AKT pathway inhibitor wortmannin inhibited colony formation. Blocking interleukin (IL)6R did not inhibit colony formation in good agreement with a lack of constitutive P-STAT3. We showed that primary cells frequently co-expressed IGF1R/IGF1 but not C-KIT/SCF or IL6R/IL6, suggesting that in vivo autonomous growth could be possible via IGF1R. Despite their similar role in clonogenic growth and shared signaling pathway, IGF1R and C-KIT had opposite prognostic values, suggesting that they were surrogate markers. Indeed, we showed that both C-KIT and IGF1R prognostic values were not independent of MMSET expression. This study highlights the autocrine role of IGF1 in myeloma cells and reinforces the interest in targeting IGF1R in IGFR1+ CD45+/− patients, such as MMSET+ patients.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is a plasma-cell malignancy that remains a fatal disease despite significant progress in its treatment over the last 10 years. This feature of MM could be related to the existence of chemotherapy-resistant myeloma stem cells. Although the myeloma stem cell type, that is, plasma cells or B cells, is still a matter of debate, recent studies have confirmed that clonogenic and tumor-initiating myeloma cells originate from plasma cells that is, CD138+ cells.1, 2 These studies reinforce the interest in identifying the survival and growth pathways and factors for myeloma ‘stem’ cells. In vitro, the study of myeloma stemness is mainly evaluated through clonogenic properties in semisolid medium. The clonogenic population allows for the establishment of human myeloma cell lines (HMCLs), confirming that this population contains cells that are able to indefinitely proliferate.1, 3 Among all of the myeloma growth factors identified to date, interleukin (IL)6, insulin-like growth factor 1 (IGF1), IL21 and members of epidermal growth factor family have been shown to sustain the growth of colonies from HMCLs and primary myeloma cells either directly or through an autocrine IGF1 loop.1, 3, 4, 5 IL6R, insulin-like growth factor 1 receptor (IGF1R) and C-KIT expression has been reported to be of prognostic value for patients treated with high-dose melphalan, with IL6R and IGF1R as adverse and C-KIT good prognostic values, respectively.6, 7, 8, 9, 10, 11, 12 The prognostic value of these three receptors could either be related to their own expression or to their restricted or overexpression by subgroups of patients marked by a prognostic value. Although IGF1R has been shown to be overexpressed in patients with t(4;14) translocation and C-KIT has been shown to be overexpressed in hyperdiploid patients, IL6R expression has not been reported to be associated with a molecular group of patients.8, 10, 11 IGF1R and C-KIT are tyrosine receptor kinases, and the signaling of both involves the AKT and ERK1,2 pathways, suggesting that the good prognostic value of C-KIT expression could be fully related to its highly restricted expression in hyperdiploid patients.4, 13, 14
In a previous study, we reported that 8 out of 32 HMCLs were able to form spontaneous colonies without the addition of serum or growth factors, suggesting that autocrine growth factors or pathways supported colony growth.15 In the present study, we investigated the involvement of IGF1, IL6 and stem cell factor (SCF) in the spontaneous colony formation of myeloma cells in a semisolid assay. We demonstrate that the IGF1 autocrine loop supports spontaneous myeloma colony growth.
Materials and methods
HMCLs and primary myeloma cells
The origin and characteristics of the HMCLs used in this study have been previously reported.16 The bone marrow and peripheral blood of patients with MM were collected after informed consent at the Nantes University Hospital.
Flow cytometry
The HMCLs were stained with a conjugated-control monoclonal antibody (mAb), the anti-IGF1R-PE mAb, the anti-JAG2-APC mAb or the anti-C-KIT-PE mAb, and fluorescence was analyzed on a FACsCalibur flow cytometer with CellQuest software (Becton Dickinson, Paris, France).
Reagents and antibodies
Allophycocyanin- and phycoerythrin-conjugated anti-CD117 (C-KIT), clone IM3638, were purchased from Beckman Coulter (Villepinte, France). The blocking anti-IGF1R mAb (AVE1642) was a gift from Aventis.17 The blocking anti-IL6R mAb (tocilizumab) was a gift from Chugai (Puteaux, France), and the blocking polyclonal goat anti-human SCF Ab was purchased from PeproTech (Neuilly sur Seine, France). Allophycocyanin-conjugated control and anti-JAG2 mAbs were purchased from R&D Systems (Lille, France). The anti-phospho-Akt (Ser473), anti-phospho-p44/42 MAPK (Thr202/Tyr204), anti-phospho-c-Kit (Tyr703) and anti-JAG2 Abs were obtained from Cell Signaling Technology, Ozyme, Saint Quentin, en Yvelines, France. Imatinib mesylate (Gleevec) was purchased from LC Laboratories (Woburn, MA, USA). Wortmannin was purchased from Sigma-Aldrich (St Quentin Fallavier, France).
Clonogenic assay
Clonogenic assays were performed in collagen semisolid medium as previously described.4, 15 This medium is currently used for the evaluation of human clonogenic hematopoietic precursors before the autologous stem cell transplant of patients diagnosed with hematological malignancies. Briefly, 103 myeloma cells were plated in 1 ml of serum- and cytokine-free, human, purified, collagen-based Stem Alpha III semisolid medium (Stem Alpha SA, Saint Genis L′Argentière, France). The experiments were performed in triplicate (330 μl per well) in four-well plates (well diameter of 1.5 cm), and the cells were grown for 21 days. The gels were collected on glass slides, dried and subjected to May-Grunwald-Giemsa staining. Colony numbers (a colony was defined as containing at least 10 cells) were counted for triplicate gels by microscopy, and are expressed as the average per 103 cells.
Western blotting
Myeloma cells (5 million cells per 10 ml) were pelleted and resuspended in lysis buffer (10 mM Tris-HCl pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 μM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin and 1% Triton X-100). After 40 min on ice, the lysates were cleared by centrifugation at 10 000 g for 30 min at 4 °C. Protein concentration was measured using bicinchoninic acid (BCA Protein assay, Pierce, Rockford, IL, USA). The cleared lysates (40–70 μg) were separated by SDS-polyacrylamide gel electrophoresis (7.5–10% acrylamide) and electrotransferred to polyvinylidene difluoride membranes. Western blot analysis was performed using standard techniques with ECL detection (Pierce Perbio Science France, Brebières, France).
Small interfering RNA transient transfections
Control nontargeted small interfering RNA (siRNA; siCt) and ON-TARGET plus SMART pool siRNA human IGF1R were purchased from Thermo Scientific (Courtaboeuf, France). siRNAs were transfected into KMM1 cells using the Lipofectamine RNAiMAX (Invitrogen, Life Technologies, Saint Aubin, France), according to the manufacturer’s instructions. Briefly, cells were plated at 1 × 106 cells per well in a six-well plate. After 24 h, siRNA (100 pmol) was transfected into the cells using Lipofectamine RNAiMAX reagent and seeded in collagen-based medium after 48 h. The gene-silencing effect was evaluated by western blot analysis.
Gene expression profiling and quantitative real-time PCR
The gene expression profiling of HMCLs was previously reported.16 We also used Affymetrix data (Santa Clara, CA, USA) from a cohort of 345 purified myeloma cells from previously untreated patients from the Arkansas Cancer Research Center (ACRC, Little Rock, AR, USA). The patients were treated with total therapy 2 that included high-dose melphalan and autologous stem cell transplantation (ASCT). These data are publicly available on the online Gene Expression Omnibus (Gene Expression Profile of Multiple Myeloma, accession number GSE2658. http://www.ncbi.nlm.nih.gov/geo/). Quantitative PCR was performed in duplicate using the TaqMan Universal PCR Master Mix (Applied Biosystems, Villebon sur Yvette, France) and the MX3005 instrument (Stratagene, Agilent Technologies, Massy, France). TaqMan gene expression assays for IGF1 (Hs00153126_m1) and RPL37a (Hs01102345_m1) were from Applied Biosystems.
Statistical analysis
Statistical analyses were performed using the Mann–Whitney test, χ2-test, Pearson test, the log-rank test and Student’s t-test with GraphPad Prism software (GraphPad, Ritme Informatique, Paris, France). COX analyses were performed with the Sigmaplot software (Systat, Ritme Informatique, Paris, France).
Results
IGF1, but not IL6, is the main clonogenic self-growth factor for myeloma cell lines
As exogenous IL6 and IGF1, the main myeloma growth factors, were able to sustain the growth of myeloma colonies, we hypothesized that autocrine loop(s) of these growth factors could be involved in spontaneous colony formation of eight HMCLs (JJN3, KARPAS 620, KMM1, L363, NCI-H929, RPMI8226, XG7 and XG11) in a serum-free, collagen-based, semisolid assay (Figure 1a and Chiron et al.15) As HMCLs co-expressed IGF1 and IGF1R (Table 1), we performed clonogenic assays in the presence of a blocking anti-IGF1R mAb.17 The anti-IGF1R mAb inhibited spontaneous colony formation (from 28 to 97%) in seven of eight HMCLs (Figure 1b). Notably, the percentage of inhibition of colony formation positively correlated with the level of IGF1R expression for all but two HMCLs, KARPAS 620 (triangle) and XG11 (square), as shown in Figure 1c. We previously reported that the ability of paracrine IL6 or IGF1 to sustain colony growth depends on the activation of the ERK and AKT pathways.4 As shown in Figure 1d, the HMCLs exhibited strong constitutive phosphorylation of both AKT and ERK. In the presence of the blocking anti-IGF1R mAb, constitutive AKT phosphorylation was fully and partly inhibited in KMM1 and JJN3, respectively. In contrast, constitutive ERK1,2 phosphorylation was not modulated upon IGF1R neutralization (Figure 1e). Both IGF1R and insulin receptor signaling are inhibited by the tyrosine phosphatase CD45, which directly dephosphorylates both receptors.17, 18 All HMCLs but XG11 did not express CD45, which agrees with the involvement of IGF1R signaling (Table 1). As the clonogenic growth of XG11 was inhibited, although only partially, by the anti-IGF1R mAb, it is possible that IGF1R signaling occurred despite the expression of CD45. Indeed, XG11 expressed a very high level of IGF1R (confirmed by flow cytometry). In contrast to IGF1, IL6 expression (Table 1) and the subsequent constitutive phosphorylation of STAT3 could not be detected in all but one HMCL, NCI-H929 (data not shown). Furthermore, clonogenic assays in the presence of 5 μg/ml of the blocking of anti-IL6R mAb Tocilizumab (Chugai), which prevents the binding of IL6 to the gp80 chain,19, 20 did not significantly affect colony number (data not shown).

Spontaneous clonogenic growth of HMCLs is mostly mediated by IGF1R autoactivation. (a) Images of colony-formation unit (CFU)-MM formation. Cells were seeded in semisolid collagen. After 3 weeks, the entire well was transferred onto a glass slide and the cells were subjected to May-Grunwald staining. (b) Inhibition of colony formation in the presence of the blocking anti-IGF1R mAb. Cells were seeded 21 days before analysis, as described in a, with or without the addition of the anti-IGF1R mAb (6 μg/ml). The colonies were counted, and the results are expressed as the percentage of inhibition of colony formation. The data represent the mean±s.d. of three independent experiments. (c) Inhibition of colony formation correlated with IGF1R expression. The IGF1R expression level (microarray data) was plotted against the percentage of inhibition of colony formation in the presence of the anti-IGF1R mAb. The square and triangle plots represent XG11 and KARPAS 620 HMCLs, respectively. Circle plots represent the remaining HMCLs. (d) Western blot analysis of constitutive AKT and ERK1/2 phosphorylation. Cells were cultured for 3 days in serum-free Roswell Park Memorial Institute medium (RPMI) 1640 supplemented with 0.5% bovine serum albumin (BSA). Equivalent amounts of cell lysate were subjected to immunoblotting with the indicated antibodies. The western blot analysis was performed as described in the Materials and Methods. (e) Constitutive AKT but not ERK phosphorylation was inhibited by neutralizing anti-IGF1RmAb. Western blot analysis of constitutive P-ERK and P-AKT in the presence of the anti-IGF1R mAb. Cells were cultured for 3 days in serum-free RPMI 1640 supplemented with 0.5% BSA, with or without IGF1R mAb (6 μg/ml). *P<0.05, **P<0.01.
Collectively, our data suggest that IGF1 but not IL6 is the main self-clonogenic growth factor for myeloma cell lines.
SCF is a clonogenic self-growth factor for a subset of myeloma cell lines
As the IGF1R-blocking mAb did not fully inhibit colony formation in several cell lines, we looked for other growth factor autocrine loops. It was previously reported that C-KIT signaling involves ERK1/2 and AKT activation.13, 21 We showed that three out of eight HMCLs, XG11, KARPAS 620 and JJN3 (50% of cells) expressed C-KIT, and addition of SCF increased the phosphorylation of both AKT and ERK1,2 in XG11 and JJN3 (Figures 2a and b). We then explored whether a SCF-C-KIT loop could be involved using an inhibitor of C-KIT signaling (imatinib mesylate or Gleevec) or a blocking anti-SCF mAb. Imatinib mesylate (10 μM) inhibited the colony formation of XG11, JJN3 and KARPAS 620 (90%±10, 70±5% and 38±2%, respectively), whereas the colony formation of two C-KIT– HMCLs, KMM1 and RPMI8226 was unaffected (Figure 2c). Similarly, neutralization of SCF with an anti-SCF mAb (500 ng/ml) significantly reduced the colony formation of XG11 and JJN3 (55±5.6% and 33±10% inhibition, respectively) but did not significantly affect that of KARPAS 620 (15±3% inhibition) or C-KIT– KMM1 (3±6% inhibition), as shown in Figure 2d, which correlates with SCF expression (Table 1). Furthermore, we showed that C-KIT was constitutively phosphorylated in XG11, which was inhibited by the blocking anti-SCF mAb but not by the anti-IGF1R mAb (Figure 2e). In the presence of both mAbs, the phosphorylation of AKT but not that of ERK1,2 was inhibited (Figure 2f), and the inhibition of colony formation induced by each mAb was additive (data not shown).

Spontaneous clonogenic growth of a subset of HMCLs is mediated by C-KIT autoactivation. (a) Flow cytometry analysis of C-KIT expression. Cell staining and fluorescence analyses were performed as described in the Materials and methods. (b) Western blot analysis of the SCF-induced kinetic phosphorylation of AKT and ERK1,2. Cells were cultured for 24 h in serum-free RPMI 1640 supplemented with 0.5% bovine serum albumin (BSA), washed and incubated with or without SCF (10 ng/ml) as indicated. (c) Imatinib inhibited colony formation of C-KIT-positive cell lines only. Clonogenic assays were performed as described in Figure 1, with or without 10 μM imatinib mesylate. The colonies were counted, and the results are expressed as the percentage of inhibition of colony formation. The data represent the mean±s.d. of three independent experiments. (d) The blocking anti-SCF mAb inhibited colony formation. XG11, JJN3, KARPAS 620 and KMM1 cells were seeded for 21 days with or without 500 ng/ml blocking anti-SCF mAb. The colonies were counted, and the results are expressed as the percentage of inhibition of colony formation. The data represent the mean±s.d. of three independent experiments. (e) Western blot analysis of constitutive P-C-KIT in the presence of the anti-IGF1R and/or anti-SCF mAbs. Cells were cultured for 24 h in serum-free RPMI 1640 supplemented with 0.5% BSA, washed and further incubated for 30 h with or without anti-IGF1R (6 μg/ml) or anti-SCF (6 μg/ml), as indicated. The western blot analysis was performed as described in Figure 1. (f) Western blot analysis of constitutive P-AKT and P-ERK in the presence of the anti-IGF1R and/or anti-SCF mAbs. Cells were cultured for 24 h in serum-free RPMI 1640 supplemented with 0.5% BSA, washed and further incubated for 30 h with or without anti-IGF1R (6 μg/ml) or anti-SCF (6 μg/ml), as indicated. The western blot analysis was performed as described in Figure 1. (g) The PI3K/AKT inhibitor wortmannin inhibited colony formation. Cells were seeded 21 days before analysis, as described in Figure 1a, with or without the addition of wortmannin (10 μM). The colonies were counted, and the results are expressed as the percentage of inhibition of colony formation. The data represent the mean±s.d. of two independent experiments performed in duplicate wells. *P<0.05, **P<0.01.
These data collectively demonstrate the involvement of an IGF1 and/or SCF loop in the spontaneous clonogenic ability of HMCLs. The heterogeneous level of constitutive P-AKT depends not only on IGF1/IGF1R and SCF/C-KIT expression level, but also on the expression of phosphatases CD45 and PTEN, which are differentially expressed across cell lines (Table 1). To confirm the direct role of PI3K/AKT pathway in the spontaneous colony formation, we performed clonogenic assays in the presence of 10 μM wortmannin, an inhibitor of PI3K/AKT pathway.14 Wortmannin inhibited colony formation of KARPAS 620, RPMI8226 and XG11 cells by 82±6%, 73±11% and 59±11%, respectively, (Figure 2g). These results confirmed the involvement of PI3K/AKT pathway in clonogenic growth.
Silencing of IGF1R, imatinib and wortmannin did not inhibit JAG2 expression
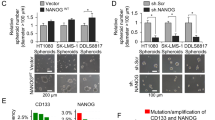
As we previously reported that JAG2 expression was required for spontaneous colony formation, we monitored JAG2 expression following inhibition of PI3K/AKT pathway by IGF1R silencing, imatinib and wortmannin.15 The silencing of IGF1R reduced the colony formation by 81±10% (Figures 3a and b, P<0.01, t-test), the constitutive phosphorylation of AKT but not the expression of JAG2 in KMM1 cells (Figure 3c). Reciprocally, P-AKT level was not decreased upon stable silencing of JAG2 (shJAG2), despite an inhibition of colony formation (Figure 3d and Chiron et al.15). Similarly, inhibition of C-KIT pathway by imatinib (20 μM) or of PI3K/AKT pathway by wortmannin (10 μM) did not induce a decrease in JAG2 expression in C-KIT+ XG11 or in C-KIT- KMM1 cells (Figure 3e).

Silencing of IGFR1, imatinib and wortmannin did not decrease JAG2 expression. (a) IGF1R silencing. KMM1 cells were transiently transfected with siControl or siRNA human IGF1R (siIGF1R) messenger RNA (mRNA) for 48 h and IGF1R expression was assessed by flow cytometry. Overlay histograms represent the IGF1R staining (thick line) over that of control staining (thin line). The specific fluorescence was expressed as the ratio of the specific mean fluorescence intensity divided by the control fluorescence (r). A representative experiment out of two is shown. (b) IGF1R silencing inhibited the spontaneous colony formation. KMM1 cells were transiently transfected with siCt or siIGF1R mRNA for 48 h, and 660 cells were seeded in two wells containing semisolid collagen. Colonies were counted after 3 weeks. The data represent the means±s.d. of four wells from two experiments. (c) IGF1R silencing did not inhibit JAG2 expression. The western blot analysis was performed 48 h after siRNA transfection as described in a. (d) JAG2 silencing did not inhibit P-AKT. Stable shJAG2 and shControl KMM1 cells15 were incubated for 48 h in serum-free RPMI 1640 medium supplemented with 0.5% bovine serum albumin (BSA). (e) Imatinib and wortmannin did not decrease the expression of JAG2. Cells were treated for 48 h with 20 μM imatinib or 10 μM wortmannin in serum-free RPMI 1640 medium supplemented with 0.5% BSA, and JAG2 expression was assessed by flow cytometry. Overlay histograms represent the JAG2 staining (thick lines) over that of control staining (thin lines). **P<0.01.
These data showed that the inhibition of PI3K/AKT pathway by IGF1R silencing, imatinib or wortmannin did not induce a decrease in JAG2 expression, despite an inhibition of colony formation.
Primary myeloma cells collected at diagnosis frequently overexpressed IGF1R, C-KIT and IGF1, but not SCF
C-KIT and IGF1R expression in myeloma cells has been previously reported.6, 7, 8, 9, 10, 11, 12 To address the expression of C-KIT/SCF, IGF1R/IGF1 and IL6R/IL6 in primary myeloma cells, we used the Amazonia database, which contains public data from 345 patients at diagnosis and was published by Arkansas University.10 C-KIT, IGF1R and IL6R were frequently expressed by 52.9%, 79.7% and 98.3% of patients, respectively (Figure 4a). Notably, only 10% of the patients neither expressed C-KIT nor IGF1R, 41% co-expressed both receptors and 49% expressed one or the other. Myeloma cells frequently expressed IGF1 (99%) but not SCF (2.3%) or IL6 (20.6%), as shown in Figure 4a. Moreover, both IGF1R and IGF1 were highly expressed by myeloma cells (Figure 4b), and IGF1 messenger RNA expression was confirmed by real-time quantitative-PCR in four samples of purified myeloma cells from patients with MM at diagnosis. All MM samples expressed high level of IGF1 messenger RNA compared with XG7 and NCI-H929 HMCLs (median value of 119-fold, n=4, P=0.002 Mann–Whitney test; Figure 4c). These data suggested that, at least at diagnosis, myeloma cells could grow through an IGF1 but not a SCF or IL6 loop.

Primary myeloma cells express IGF1R and IGF1. (a) Frequency of expression of C-KIT/SCF, IGF1R/IGF1 and IL6R/IL6 (absent/present) was assessed by microarray in 345 samples (public database from Arkansas University) from patients at diagnosis, as previously described.16 (b) Expression level of IGF1R and IGF1 was assessed by microarray as in a. Only samples with positive calls were included. (c) Expression of IGF1 messenger RNA was assessed by real-time quantitative-PCR in HMCLs and primary samples. Data were normalized with XG7 as calibrator. MM1-3, bone marrow myeloma cells; MM4, peripheral blood myeloma cells. (d) The frequency of expression of C-KIT, IGF1R and IL6R (absent/present) was compared in MMSET+ and MMSET− patients by microarray. (e) The level of expression of C-KIT, IGF1R and IL6R (only positive calls were included) was compared in MMSET+ and MMSET− patients. **P<0.01, ***P<0.001.
The favorable prognosis of C-KIT expression is linked to a lack of MMSET expression
The expression of C-KIT or IGF1R has been shown to be associated with good and bad prognoses for patients with MM, respectively. These opposing prognostic values are surprising because both tyrosine kinase receptors mediate clonogenic growth and similar activation of the AKT and ERK1,2 pathways, suggesting that they could be surrogate markers. Indeed, patients with the t(4;14) translocation overexpress MMSET and have a poor outcome, overexpress IGF1R and underexpress C-KIT.10, 11 MMSET was expressed by 15% of patients. C-KIT was expressed by 58.4% of MMSET– patients and 21% of MMSET+ patients (P<0.0001, χ2-test, Figure 4d). In contrast, IGF1R was expressed by 76% and 96% of MMSET– and MMSET+ patients, respectively, (P=0.0001, χ2-test, Figure 4d). The frequency of expression of IL6R was similar in both subsets of patients. However, intensity-level analyses showed that MMSET+ patients significantly overexpressed IGF1R and IL6R (P<0.0001) but underexpressed C-KIT compared with MMSET– patients (P=0.01), as shown in Figure 4e.
We re-evaluated the prognosis of C-KIT, IGF1R in comparison with that of MMSET. As previously reported and shown in Figure 5a, the expression of C-KIT was of favorable prognostic value (P=0.0228) whereas that of IGF1R or MMSET was of adverse prognostic value (P=0.046 and P<0.0001, respectively). We next performed the same analysis in the MMSET− cohort of patients (n=293) and found that the expression of C-KIT was not significantly linked to patient outcome (P=0.46) and that of IGF1R retained its trend (P=0.1, Figure 5b). In the MMSET+ group, the expression of none of these genes was significantly linked to survival (Figure 5c). A multivariate analysis showed that MMSET was the only independent prognostic value (P<0.001, Table 2).

IGF1R and C-KIT are not prognostic values for MMSET− patients. Kaplan–Meyer graphs were constructed for the whole population (a), the MMSET− population (b) and the MMSET+ (c) population. The log-rank test was used for survival statistical analysis.
Discussion
In this study, we identified the growth factors involved in myeloma self-clonogenic growth using eight HMCLs in a serum-free collagen assay. We showed that colony growth was inhibited upon neutralization of IGF1R in seven HMCLs or upon neutralization of SCF in two HMCLs. Notably, the level of inhibition was significantly and positively correlated to IGF1R expression. In contrast, neutralization of IL6 did not significantly modulate the clonogenic growth of any HMCLs. We previously reported that colony formation in the presence of added growth factors is related to the ability of the growth factors to induce ERK1,2 and AKT phosphorylation.4 Interestingly, we showed that HMCLs that were able to produce spontaneous clones had constitutive phosphorylation of both ERK1,2 and AKT. However, neutralization of IGF1R or SCF inhibited the constitutive AKT phosphorylation but did not modulate that of ERK1,2. All of the analyzed HMCLs, except XG11, had a RAS mutation, which could explain the constitutive phosphorylation of ERK1,2.16 These data show that AKT phosphorylation is associated with self-colony formation and strongly suggest that self-colony formation is dependent on AKT phosphorylation. Indeed, we showed that the PI3K/AKT pathway inhibitor wortmannin inhibited colony formation. Oncogenic activation of AKT has been widely described in myeloma and has been identified as a potential therapeutic target.22, 23 IGF1 and insulin activate the AKT pathway, whereas CD45 decreases this signaling by dephosphorylating both IGF1R and INSR.14 Except for XG11, all HMCLs demonstrating self-colony formation through the IGF1 loop did not express CD45. Concerning XG11, its very strong expression of IGF1R could explain why an IGF1 loop is still efficient despite the expression of CD45 (Table 1 and Figure 1). The IGF1 autocrine loop has been shown to mediate both IL21- and FGF-induced myeloma colony formation.4, 5 Moreover, an IGF1 loop is also involved in the IL6-induced growth of myeloma cells in liquid culture.6 Recently, Barretina et al.24 reported that among numerous cancer cell lines, myeloma cells (mainly CD45– cell lines) were the most sensitive to the inhibition of IGF1R signaling. Considering all of these results, it appears that a spontaneous or induced IGF1 loop is the major growth pathway for myeloma cells.
The second self-clonogenic growth factor that we identified in this study was SCF. Blocking SCF/C-KIT with a neutralizing mAb or imatinib inhibited the colony formation of JJN3 and XG11. Similar results were obtained with dasatinib (data not shown). XG11 displays basal phosphorylation of C-KIT, which is inhibited by anti-SCF but not by anti-IGF1R. The combination of anti-IGF1R and anti-SCF inhibited basal AKT phosphorylation in JJN3 (Figure 2) and XG11 (data not shown). Of note, both JJN3 and XG11 were dependent on IGF1R and C-KIT for growth, and complete inhibition of AKT phosphorylation was reached only with the combination of both mAbs. KARPAS 620 was the only HMCL able to form self-colonies that was not inhibited by anti-IGF1R or anti-SCF. However, colony formation was inhibited by imatinib, suggesting the SCF-independent self-activation of C-KIT in this HMCL, which requires further investigation.
We showed that inhibition of IGF1R, C-KIT or of the PI3K/AKT pathway did not decrease the level of JAG2 expression, and we previously demonstrated that expression of JAG2 was required for spontaneous colony formation and in vivo tumor growth.15 These data collectively showed that self-colony formation depends on both JAG2 expression and IGF1R/C-KIT activation of AKT pathway.
Although the ERK and AKT pathways are similarly activated by IGF1 or SCF in HMCLs,13, 14 the IGF1 loop was more frequently involved. Indeed, most HMCLs express IGF1R/IGF1 but rarely express C-KIT/SCF. As HMCLs, primary myeloma cells commonly expressed IGF1 (80%) but not SCF (2%), and 90% of patients expressed IGF1R and/or C-KIT. Both receptors are tumor markers in MM; although they are absent in normal plasma cells, they are expressed in monoclonal gammopathy of undetermined significance (MGUS) and MM. However, despite sharing a signaling pathway that involves AKT and ERK1/2, these two tyrosine kinase receptors provide opposite prognostic values for patients. IGF1R expression at diagnosis is associated with a reduced survival rate and that of C-KIT is associated with a longer survival rate.7, 8, 9 To understand how the expression of a tyrosine kinase receptor that supports the self-clonogenic growth of the tumor could be a benefit for patients, we re-evaluated the prognosis of C-KIT expression as well as that of IGF1R in comparison with that of MMSET. Indeed, MMSET overexpression, which is due to t(4;14) chromosomal translocation, is known to have a very strong adverse prognostic value in myeloma.10, 16 C-KIT and IGF1R expression was significantly different in MMSET+ and MMSET− patients, suggesting that their prognostic values could be surrogate markers. Indeed, C-KIT expression was dependent on that of MMSET, suggesting that the favorable prognosis was only related to the lack of MMSET expression. In contrast, IGF1R retained a trend. Expression of C-KIT has been reported to be less frequent in patients with relapse compared with patients at diagnosis. As serial analyses of C-KIT expression during MGUS/MM transition or diagnosis/relapse are not available, we cannot conclude whether C-KIT is lost or C-KIT+ patients relapse less frequently than C-KIT− patients. Recent work has shown that C-KIT might act as an anchor molecule for monoclonal plasma cells in bone marrow niches, suggesting that C-KIT+ plasma cells could be more dependent on the microenvironment for growth than IGF1R+ plasma cells, which produce IGF1 but not SCF.25 C-KIT is expressed in approximately one-third of patients with MGUS (27%, n=463, N Robillard; unpublished data) or MM.8, 26 It would be interesting to study the transformation rate and frequency of C-KIT+ compared with C-KIT− MGUS in both prospective and retrospective studies to define whether C-KIT expression is predictive, or not, of the transformation of MGUS into MM.
The IGF1R or/and C-KIT growth pathways are not myeloma specific. Numerous cancer cell types, such as melanoma or acute leukemia, use the IGF1R or C-KIT pathways for growth.27, 28 Our in vitro data indicate that both IGF1R and C-KIT tyrosine receptors mediate the growth of myeloma cells, suggesting that targeting these tyrosine receptors or their downstream signaling pathways controlled by AKT should be efficient to block the self-renewal of MM cells. Myeloma cells widely express IL6R, and IL6 is a major myeloma growth and survival factor.29 However, we did not find any HMCL that uses an IL6 loop for clonogenic growth. These findings are in agreement with the non-frequent expression of IL6 messenger RNA by primary cells (26%) and HMCLs (5 out of 32, 16%). These data strongly argue in favor of a paracrine role for SCF or IL6 and an autocrine role for IGF1.
Clinical trials targeting IGF1R or C-KIT have been reported for MM. Despite some positive responses, Moreau et al.30 and Lacy et al.31 reported that the use of anti-IGF1R mAbs with dexamethasone or bortezomib was not very efficient. This less than expected efficiency, which has compromised further developments, could be related to a real inefficiency or a lack in patient selection with respect to IGF1R and CD45 expression.32 Nevertheless, IGF1R remains a target in oncology, and IGF1R tyrosine inhibitors are under development. We may expect that the use of tyrosine inhibitors rather than mAbs could be more efficient owing to their lower specificity, which could allow for the inhibition of INSR/IGF1R hybrids as well. Concerning C-KIT, imatinib has been assessed in refractory C-KIT+ and C-KIT− MM patients and does not show effective treatment.33 Despite these disappointing results, the present study highlights that IGF1 is an essential growth factor for myeloma cells. Preclinical in vitro and in vivo studies have shown that blocking IGF1R with mAbs or inhibitors synergizes with bortezomib and overcomes bortezomib resistance.34, 35 New drugs, new associations and/or the careful selection of patients could improve the currently insufficient clinical results. For example, targeting IGF1R in combination with bortezomib could be of particularly interest for MMSET+ patients, who overexpress IGF1R. In summary, our results identify the crucial role of autocrine IGF1 in self-colony formation and emphasize the interest to target AKT, the shared downstream signaling target of IGF1R and C-KIT, for the treatment of MM.
Accession codes
References
Chiron D, Surget S, Maiga S, Bataille R, Moreau P, Le Gouill S et al. The peripheral CD138+ population but not the CD138- population contains myeloma clonogenic cells in plasma cell leukaemia patients. Br J Haematol 2012; 156: 679–683.
Kim D, Park CY, Medeiros BC, Weissman IL . CD19-CD45 low/− CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012; 26: 2530–2537.
Hitzler JK, Martinez-Valdez H, Bergsagel DB, Minden MD, Messner HA . Role of interleukin-6 in the proliferation of human multiple myeloma cell lines OCI-My 1 to 7 established from patients with advanced stage of the disease. Blood 1991; 78: 1996–2004.
Collette M, Descamps G, Pellat-Deceunynck C, Bataille R, Amiot M . Crucial role of phosphatase CD45 in determining signaling and proliferation of human myeloma cells. Eur Cytokine Netw 2007; 18: 120–126.
Menoret E, Maiga S, Descamps G, Pellat-Deceunynck C, Fraslon C, Cappellano M et al. IL-21 stimulates human myeloma cell growth through an autocrine IGF-1 loop. J Immunol 2008; 181: 6837–6842.
Sprynski AC, Hose D, Caillot L, Reme T, Shaughnessy JD Jr, Barlogie B et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009; 113: 4614–4626.
Bataille R, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P . CD221 (IGF-1R) is aberrantly expressed in multiple myeloma, in relation to disease severity. Haematologica 2005; 90: 706–707.
Bataille R, Pellat-Deceunynck C, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P . CD117 (c-kit) is aberrantly expressed in a subset of MGUS and multiple myeloma with unexpectedly good prognosis. Leuk Res 2008; 32: 379–382.
Chng WJ, Gualberto A, Fonseca R . IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia 2006; 20: 174–176.
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S et al. The molecular classification of multiple myeloma. Blood 2006; 108: 2020–2028.
Mateo G, Montalban MA, Vidriales MB, Lahuerta JJ, Mateos MV, Gutierrez N et al. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. J Clin Oncol 2008; 26: 2737–2744.
Schmidt-Hieber M, Perez-Andres M, Paiva B, Flores-Montero J, Perez JJ, Gutierrez NC et al. CD117 expression in gammopathies is associated with an altered maturation of the myeloid and lymphoid hematopoietic cell compartments and favorable disease features. Haematologica 2011; 96: 328–332.
Pandiella A, Carvajal-Vergara X, Tabera S, Mateo G, Gutierrez N, San Miguel JF . Imatinib mesylate (STI571) inhibits multiple myeloma cell proliferation and potentiates the effect of common antimyeloma agents. Br J Haematol 2003; 123: 858–868.
Descamps G, Pellat-Deceunynck C, Szpak Y, Bataille R, Robillard N, Amiot M . The magnitude of Akt/phosphatidylinositol 3′-kinase proliferating signaling is related to CD45 expression in human myeloma cells. J Immunol 2004; 173: 4953–4959.
Chiron D, Maiga S, Descamps G, Moreau P, Le Gouill S, Marionneau S et al. Critical role of the NOTCH ligand JAG2 in self-renewal of myeloma cells. Blood Cells Mol Dis 2012; 48: 247–253.
Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica 2011; 96: 574–582.
Descamps G, Wuilleme-Toumi S, Trichet V, Venot C, Debussche L, Hercend T et al. CD45neg but not CD45pos human myeloma cells are sensitive to the inhibition of IGF-1 signaling by a murine anti-IGF-1R monoclonal antibody, mAVE1642. J Immunol 2006; 177: 4218–4223.
Kulas DT, Freund GG, Mooney RA . The transmembrane protein-tyrosine phosphatase CD45 is associated with decreased insulin receptor signaling. J Biol Chem 1996; 271: 755–760.
Nishimoto N, Kishimoto T, Yoshizaki K . Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann Rheum Dis 2000; 59 (Suppl 1): i21–i27.
Nishimoto N, Sasai M, Shima Y, Nakagawa M, Matsumoto T, Shirai T et al. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000; 95: 56–61.
Montero JC, Lopez-Perez R, San Miguel JF, Pandiella A . Expression of c-Kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica 2008; 93: 851–859.
Steinbrunn T, Stuhmer T, Gattenlohner S, Rosenwald A, Mottok A, Unzicker C et al. Mutated RAS and constitutively activated Akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood 2011; 117: 1998–2004.
Mahindra A, Cirstea D, Raje N . Novel therapeutic targets for multiple myeloma. Future Oncol 2010; 6: 407–418.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603–607.
Paiva B, Perez-Andres M, Vidriales MB, Almeida J, de las Heras N, Mateos MV et al. Competition between clonal plasma cells and normal cells for potentially overlapping bone marrow niches is associated with a progressively altered cellular distribution in MGUS vs myeloma. Leukemia 2011; 25: 697–706.
Ocqueteau M, Orfao A, Almeida J, Blade J, Gonzalez M, Garcia-Sanz R et al. Immunophenotypic characterization of plasma cells from monoclonal gammopathy of undetermined significance patients. Implications for the differential diagnosis between MGUS and multiple myeloma. Am J Pathol 1998; 152: 1655–1665.
Smalley KS, Nathanson KL, Flaherty KT . Genetic subgrouping of melanoma reveals new opportunities for targeted therapy. Cancer Res 2009; 69: 3241–3244.
Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010; 95: 415–423.
Zhang XG, Gaillard JP, Robillard N, Lu ZY, Gu ZJ, Jourdan M et al. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood 1994; 83: 3654–3663.
Moreau P, Cavallo F, Leleu X, Hulin C, Amiot M, Descamps G et al. Phase I study of the anti insulin-like growth factor 1 receptor (IGF-1R) monoclonal antibody, AVE1642, as single agent and in combination with bortezomib in patients with relapsed multiple myeloma. Leukemia 2011; 25: 872–874.
Lacy MQ, Alsina M, Fonseca R, Paccagnella ML, Melvin CL, Yin D et al. Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulinlike growth factor type 1 Receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. J Clin Oncol 2008; 26: 3196–3203.
Bataille R, Robillard N, Pellat-Deceunynck C, Amiot M . A cellular model for myeloma cell growth and maturation based on an intraclonal CD45 hierarchy. Immunol Rev 2003; 194: 105–111.
Dispenzieri A, Gertz MA, Lacy MQ, Geyer SM, Greipp PR, Rajkumar SV et al. A phase II trial of imatinib in patients with refractory/relapsed myeloma. Leuk Lymphoma 2006; 47: 39–42.
Descamps G, Gomez-Bougie P, Venot C, Moreau P, Bataille R, Amiot M . A humanised anti-IGF-1R monoclonal antibody (AVE1642) enhances Bortezomib-induced apoptosis in myeloma cells lacking CD45. Br J Cancer 2009; 100: 366–369.
Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012; 120: 3260–3270.
Acknowledgements
We thank Dr T Guillot (Chugai, Neuilly, France) and Dr C Venot (Aventis, France) for providing us with the blocking anti-IL6R mAb (gp80) Tocilizumab and the blocking anti-IGF1R AVE1642 mAb, respectively. This work was supported by grants from Cancéropôle Grand-Ouest (AO2009 Cellules souches).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
DC performed the research and participated in the design of the study and the writing of the manuscript. SM, SS, GD, PGB, ST and NR performed the research and reviewed the manuscript. PM and SLG provided primary samples and reviewed the manuscript. RB and MA participated in the design of the study and the writing of the manuscript. CPD designed the study and wrote the manuscript.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chiron, D., Maïga, S., Surget, S. et al. Autocrine insulin-like growth factor 1 and stem cell factor but not interleukin 6 support self-renewal of human myeloma cells. Blood Cancer Journal 3, e120 (2013). https://doi.org/10.1038/bcj.2013.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2013.18
Keywords
This article is cited by
-
Combined targeting of MEK and the glucocorticoid receptor for the treatment of RAS-mutant multiple myeloma
BMC Cancer (2020)
-
Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma
International Journal of Hematology (2016)
-
Silencing stem cell factor attenuates stemness and inhibits migration of cancer stem cells derived from Lewis lung carcinoma cells
Tumor Biology (2016)
