
Abstract
The use of ginseng extract as an adjuvant for cancer treatment has been reported in both animal models and clinical applications, but its molecular mechanisms have not been fully elucidated. Mitomycin C (MMC), an anticancer antibiotic used as a first- or second-line regimen in the treatment for non-small cell lung carcinoma (NSCLC), causes serious adverse reactions when used alone. Here, by using both in vitro and in vivo experiments, we provide evidence for an optimal therapy for NSCLC with total ginsenosides extract (TGS), which significantly enhanced the MMC-induced cytotoxicity against NSCLC A549 and PC-9 cells in vitro when used in combination with relatively low concentrations of MMC. A NSCLC xenograft mouse model was used to confirm the in vivo synergistic effects of the combination of TGS with MMC. Further investigation revealed that TGS could significantly reverse MMC-induced S-phase cell cycle arrest and inhibit Rad51-mediated DNA damage repair, which was evidenced by the inhibitory effects of TGS on the levels of phospho-MEK1/2, phospho-ERK1/2 and Rad51 protein and the translocation of Rad51 from the cytoplasm to the nucleus in response to MMC. In summary, our results demonstrate that TGS could effectively enhance the cytotoxicity of MMC against NSCLC cells in vitro and in vivo, thereby revealing a novel adjuvant anticancer mechanism of TGS. Combined treatment with TGS and MMC can significantly lower the required concentration of MMC and can further reduce the risk of side effects, suggesting a better treatment option for NSCLC patients.
Similar content being viewed by others

Introduction
Lung cancer is among the leading causes of cancer death among males in both developed and developing countries, and it has surpassed breast cancer among females in developed countries. Approximately 1.8 million new lung cancer cases were diagnosed in 2012, accounting for approximately 13% of total cancer diagnoses1. Non-small cell lung carcinoma (NSCLC) accounts for 80% of lung cancers2. Chemotherapy is the main therapy for NSCLC patients, however, toxic side effects limit its clinical application to a great extent3,4,5. Mitomycin C (MMC) is an anticancer agent that causes intrastrand DNA crosslinks (ICLs), which are among the most severe types of DNA damage and block DNA processes such as transcription and replication6. Currently, MMC is being extensively used as a chemotherapeutic agent in the clinic for the treatment of a variety of cancers, including stomach, breast, pancreas, colon and bladder cancer7,8,9. MMC has been formulated to treat NSCLC as a single agent and can be combined with other chemotherapy agents for advanced NSCLC treatment in the form of a first- or second-line regimen. However, it exhibits prolonged suppression of the bone marrow. Therefore, the role of MMC in the treatment of NSCLC needs to be redefined10,11. At present, NSCLC remains a threat to human health, with an overall 5-year survival rate of only 18.2%12.
Many natural products have anticancer effects and can enhance the cytotoxicity of chemotherapy agents13,14. It is a common strategy to use combinations of natural products and chemotherapy agents as anticancer treatment to overcome drug resistance and reduce toxicity due to the safety and efficacy of natural products15,16; however, the underlying mechanisms of the antitumor effects of most natural products are still unknown.
Ginseng (Panax ginseng C. A. Mey) has been widely used in Asian countries for thousands of years17. The effective components of ginseng are multiple ginsenosides, including more than 100 types of protopanaxadiol (PPD) and protopanaxatriol (PPT)-type ginsenosides18. The pharmacological activities and pharmacokinetic behaviors of these ginsenosides have been identified in many studies19,20,21. Ginseng and Shenmai injection (ginseng-derived Chinese medicine) are prescribed as herbal medicines for cancer patients in China; however, their underlying adjuvant anticancer mechanisms are still elusive.
This study was designed with the aim of elucidating the signaling pathways associated with promoting cell death in NSCLC by total ginsenosides extract (TGS) in combination with MMC. In brief, TGS significantly upregulated the anticancer effect of MMC in A549 and PC-9 cells in a synergistic manner. The underlying mechanism of this synergistic efficacy involved the MEK1/2-ERK1/2-Rad51 pathway, which is responsible for the poor efficacy of long-term MMC treatment. The synergistic efficacy of TGS combined with MMC was further confirmed in a xenograft model in vivo, suggesting a novel strategy for combination therapy for NSCLC patients.
Materials and methods
Chemicals and reagents
Total ginsenosides extract and ginsenoside monomers (purity 98%) were purchased from the College of Chemistry of Jilin University (Changchun, China). The ginsenosides content (Rb1 11.2%, Rb2 11.1%, Rc 10.5%, Rd 7.7%, Re 8.9%, Rf 0.9%, Rg1 3.3%, Rg2 1.4%, and Rh1 0.2%) in the total ginsenosides extract was determined using a liquid chromatography-mass spectrometry method validated in our laboratory21. TGS was dissolved in RPMI-1640 medium at a concentration of 100 mg/mL. Ginsenoside monomers were dissolved in DMSO at a concentration of 100 mmol/L. Diol-type ginsenosides and triol-type ginsenosides were dissolved in DMSO at concentrations of 0.05 mg/mL and 0.025 mg/mL, respectively. MMC was purchased from Bomei Biotechnology (Hefei, China) and dissolved in DMSO at concentrations of 0.25, 0.5, 1 and 2 mg/mL. DMSO was purchased from Sigma-Aldrich (St Louis, MO, USA). The MEK1/2 inhibitor U0126-EtOH was purchased from Selleckchem (Houston, USA). Lipofectamine iMAX, Rad51 siRNA (VHS40454) and control siRNA (medium GC) were purchased from Life Technologies (Shanghai, China). The FragELTM DNA Fragmentation Detection Kit (QIA39) was purchased from Merck Millipore (Massachusetts, USA). Cell Counting Kit 8 (CCK-8) was purchased from Dojindo (Kumamoto, Japan). RPMI-1640 and Opti-MEM medium were purchased from Gibco (California, USA). RIPA lysis buffer, Hoechst 33342, Triton X-100, PMSF, BCA protein assay kit, and nuclear and cytoplasmic protein extraction kit were purchased from Beyotime Biotechnology (Shanghai, China).
Cell culture
Human non-small cell lung cancer cell lines A549 and PC-9 were obtained from the American Type Culture Collection (ATCC) (Maryland, USA) and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C with 5% CO2. All reagents used in cell culture were from Life Technologies (Shanghai, China).
Cell proliferation assay
Cells in 96-well plates at 80% confluence were treated with 0.25, 0.5 or 0.75 mg/mL TGS and 0.25, 0.5 or 0.75 μg/mL MMC alone or in combination for 48 h. Cell viability was measured using the CCK-8 assay according to the manufacturer's instructions. Absorbance at 450 nm was measured using a BioTek Synergy H1 Hybrid Reader (Vermont, USA).
Hoechst staining
Cells in 6-well plates at 80% confluence were treated with 0.5 or 1 mg/mL TGS and 0.5 or 1 μg/mL MMC alone or in combination for 24 h, after which the medium was discarded and fresh medium containing 10 μg/mL Hoechst 33342 was added for further 30 min at 37 °C. Then, the medium was discarded, and the cells were gently washed three times with ice-cold PBS. The cells were subsequently observed with a Leica DMI3000B fluorescence microscope (Bensheim, Germany).
siRNA transfection
All transfections were performed using Lipofectamine iMAX according to the manufacturer's instructions. Cells were transfected with Rad51-specific or negative control siRNAs (GenePharma Inc, Shanghai, China) at a concentration of 20 nmol/L for 48 h before the assays. The sequence of the siRNA duplex for RAD51 is as follows: sense strand: 5′-CCACCAGACCCAGCUCCUUUAUCAA-3′, antisense strand: 5′-UUGAUAAAGGAGCUGGGUCUGGUGG-3′.
Apoptosis assay
Cells were treated with 0.75, 1.25 and 1.5 mg/mL TGS and 0.75, 1.25 and 1.5 μg/mL MMC for 24 h, washed with ice-cold PBS and fixed successively with 4% formaldehyde for 20 min at 4 °C and 75% ethanol for 2 h at -20 °C. The apoptotic cells were labeled using a FragELTM DNA Fragmentation Detection Kit. The labeled cells (1×104) were analyzed using a BD Biosciences FACSCalibur flow cytometer (San Jose, CA, USA).
Western blot analysis
Cells were collected and lysed in ice-cold RIPA lysis buffer containing 2 mmol/L PMSF. The protein concentrations in the lysates were quantified using a BCA Protein Assay Kit. Cell lysates containing 60 μg of protein were subjected to SDS-PAGE using 8%–12% gradient polyacrylamide gels (Bis-Tris Midi Gel, Life Technologies, USA) and analyzed by immunoblotting with antibodies to the corresponding proteins. The same blots were probed with GAPDH antibody as the loading control. Protein expression was quantified by densitometry using Image Lab software.
The primary antibodies (dilution ratio: 1:1000) anti-caspase 3, anti-caspase 7, anti-caspase 8, anti-PARP, anti-Bcl-xL, anti-Bak, anti-Bax, anti-Bid, anti-Bad and anti-γ-H2AX and horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG secondary antibodies (1:5000) were purchased from Cell Signaling Technology (Danvers, MA, USA), and anti-Rad51 (G-9) antibody (1:200) was from Santa Cruz Biotechnology (Dallas, MA, USA), and anti-GAPDH antibody (1: 1000) was from Bioworld Technology (Nanjing, China).
Cell cycle analysis
Cells were starved for 12 h in serum-free medium and treated with TGS or MMC alone or in combination for further 24 h, after which the cells were successively fixed with 4% formaldehyde for 20 min at 4 °C and 75% ethanol for 2 h at -20 °C. Then, the cells were stained with propidium iodide and detected using a BD Biosciences FACSCalibur flow cytometer (San Jose, CA, USA).
Quantitative real-time PCR
Extraction of total RNA was performed using the RNAiso Plus reagent (TaKaRa Biotechnology Co, Ltd, Dalian, China) according to the manufacturer's protocol. The concentration of RNA was determined by measuring the absorbance at 260 and 320 nm. Complementary DNA was generated from 500 ng of total RNA using SuperScript II Reverse Transcriptase (Life Technologies). Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was carried out using SYBR green PCR master mix (TaKaRa Biotechnology) in a reaction volume of 15 μL. Real-time PCR was performed using a Bio-Rad C1000TM Thermal Cycler (Carlsbad, CA, USA). The annealing was performed at 60 °C for 30 s. Relative gene expression analysis was performed using the 2(−ΔΔCt) method with GAPDH as the internal control. The primer sequences are shown in Supplementary Data Table 1 (forward 5′–3′, reverse 5′–3′).
Immunofluorescence
Cells were cultured in 0.17 mm glass-bottom dishes and treated with the indicated agents for the indicated times, after which the cells were fixed with 4% formaldehyde for 20 min at 4 °C, permeabilized with PBS containing 0.2% Triton X-100 for 20 min at room temperature, blocked with 5% w/v BSA in PBS/T (PBS containing 0.1% Tween-20) for 1 h at 37 °C and then incubated with primary antibodies overnight at 4 °C and washed 4 times with PBS/T. The cells were further incubated with fluorescence-conjugated secondary antibodies and Hoechst 33342 for 1 h in the dark, washed 4 times with PBS/T and observed with a Zeiss LSM 700 confocal fluorescence microscope (Oberkochen, Germany).
Animal treatments
BALB/c nude mice (specific pathogen-free (SPF) grade, 6 weeks old, 17–18 g), obtained from Shanghai Laboratory Animal Center, CAS (SLAC, Shanghai, China), were injected at the base of the forelimbs with A549 cells (5×106) growing at log phase. Mice were maintained in an SPF-grade animal room (temperature 23±2 °C, humidity 50%–60%, 12-h light-dark cycle) with free access to food and water. All animal studies were approved by the Animal Ethics Committee of China Pharmaceutical University and have been carried out in accordance with the Declaration of Helsinki. Tumor sizes were measured daily with a Vernier caliper and calculated using the following formula: tumor volume (mm3)=length×width×width/2. The mice were randomly divided into 6 groups according to their tumor volumes on the day before treatment. The initial average tumor volume in each group was approximately 40 mm3. Each group was treated as follows: Control, mice were treated with saline; TGS, mice were treated with 40 mg/kg of TGS dissolved in saline; MMC, mice were treated with 500 μg/kg of MMC dissolved in saline; MT (TGS/MMC), mice were treated with (40 mg/kg of TGS and 500 μg/kg of MMC) dissolved in saline. All agents were intraperitoneally injected once every day for two weeks.
Sample collection and histologic evaluation
After the mice were euthanized, tumors were immediately removed and weighed, and a small part of the tumors was fixed in 4% formaldehyde, embedded in paraffin, sectioned at 5 μm, and then stained with hematoxylin-eosin (HE), TUNEL and Ki67 accordingly. The tissue sections were observed with a Leica DMI3000B fluorescence microscope (Bensheim, Germany).
Statistical analyses
The data are representative of three independent experiments and are expressed as the mean±standard error of the mean (SEM). Prism 6.0 statistical software was used for data analyses. Two-tailed Student's t-test was used to determine statistical significance. P<0.05 was considered statistically significant.
Results
TGS synergizes with MMC to induce apoptosis in NSCLC cells
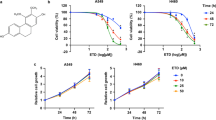
To test the regulation of TGS on cell death induced by chemotherapy agents, we co-treated A549 cells with TGS and several chemotherapy agents. The results showed that TGS synergized with MMC in inducing cell death (Figure 1A and Figure S1). CI (combination index) calculated according to Chou22, and detection of apoptosis by Hoechst and TUNEL (TdT-mediated dUTP nick-end labeling) staining confirmed the synergistic efficacy of the two agents (Figure 1B, 1C). The combination treatment increased the expression of caspase family proteins and enhanced their activation via cleavage (Figure 1D), as well as decreased the expression of anti-apoptotic protein Bcl-xL of the Bcl-2 family and increased the expression of pro-apoptotic proteins Bax, Bad, Bak and Bid (Figure 1E). The increase in cell death was significantly rescued by the pan-caspase inhibitor Z-VAD-FMK, suggesting that the observed cell death was mostly due to apoptosis (Figure 1F). We further tested the cytotoxicity of TGS on PC-9 cells, and TGS exhibited a synergistic effect on chemotherapy agent-induced apoptosis (Figure S2). These data suggest that TGS promotes caspase-dependent apoptotic cell death induced by MMC.

TGS synergizes with MMC to induce apoptosis in NSCLC A549 cells. (A) Survival curve illustrating the viability of A549 cells treated with the indicated concentrations of TGS (mg/mL) and MMC (μg/mL) alone or in combination (MT, MMC/TGS, TGS:MMC=1:1) for 48 h. The data are expressed as the cell inhibition rate relative to the untreated controls and represented as the mean±SEM (n=6). The CI (combination index) was calculated according to Chou22. Briefly, the IC50 of TGS, MMC and their combination were estimated according to their cytotoxicity and then  . (B) Analysis of Hoechst staining of cells treated with 0.5 and 1 mg/mL TGS (TGS 0.5, TGS 1) and 0.5 and 1 μg/mL MMC (MMC 0.5, MMC 1) alone or in combination (MT 0.5, MT 1, TGS:MMC=1:1) for 24 h. The experiment was performed in triplicate, and representative images are shown. Scale bar, 50 μm. (C) Analysis of TUNEL staining. The cells were treated with TGS (mg/mL) and MMC (μg/mL) alone or in combination (T0.75, T1.25 and T1.5 represent 0.75, 1.25 and 1.5 mg/mL of TGS, respectively; M0.75, M1.25 and M1.5 represent 0.75, 1.25 and 1.5 μg/mL of MMC, respectively; MT, TGS/MMC, TGS:MMC=1:1) for 24 h. Cell apoptosis was determined by TUNEL assay. (D, E) Immunoblot analysis of proteins involved in the cell apoptosis pathway. The cells were treated with the indicated agents for 24 h (T0.5 and T1 represent 0.5 mg/mL and 1 mg/mL of TGS, respectively; M1 and M2 represent 1 μg/mL and 2 μg/mL of MMC, respectively; MT1 represents 0.5 mg/mL TGS+1 μg/mL MMC; MT2 represents 1 mg/mL TGS+2 μg/mL MMC, the same as that below). GAPDH was blotted as the loading control. (F) Effect of the caspase inhibitor, Z-VAD-FMK, on the viability of cells treated with the indicated concentrations of TGS and MMC alone or in combination for 48 h. (T0.25, T0.5 and T0.75 represent 0.25, 0.5 and 0.75 mg/mL of TGS, respectively; M0.25, M0.5 and M0.75 represent 0.25, 0.5 and 0.75 μg/mL of MMC, respectively; MT0.25, MT0.5, and MT0.75 represent 0.25 mg/mL TGS+0.25 μg/mL MMC, 0.5 mg/mL TGS+0.5 μg/mL MMC, and 0.75 mg/mL TGS+0.75 μg/mL MMC, respectively). The date are represented as the mean±SEM (n=6). The represented cell viabilities are relative to that of each control, *P<0.05, **P<0.01 vs no Z-VAD-FMK. Student's t-test.
. (B) Analysis of Hoechst staining of cells treated with 0.5 and 1 mg/mL TGS (TGS 0.5, TGS 1) and 0.5 and 1 μg/mL MMC (MMC 0.5, MMC 1) alone or in combination (MT 0.5, MT 1, TGS:MMC=1:1) for 24 h. The experiment was performed in triplicate, and representative images are shown. Scale bar, 50 μm. (C) Analysis of TUNEL staining. The cells were treated with TGS (mg/mL) and MMC (μg/mL) alone or in combination (T0.75, T1.25 and T1.5 represent 0.75, 1.25 and 1.5 mg/mL of TGS, respectively; M0.75, M1.25 and M1.5 represent 0.75, 1.25 and 1.5 μg/mL of MMC, respectively; MT, TGS/MMC, TGS:MMC=1:1) for 24 h. Cell apoptosis was determined by TUNEL assay. (D, E) Immunoblot analysis of proteins involved in the cell apoptosis pathway. The cells were treated with the indicated agents for 24 h (T0.5 and T1 represent 0.5 mg/mL and 1 mg/mL of TGS, respectively; M1 and M2 represent 1 μg/mL and 2 μg/mL of MMC, respectively; MT1 represents 0.5 mg/mL TGS+1 μg/mL MMC; MT2 represents 1 mg/mL TGS+2 μg/mL MMC, the same as that below). GAPDH was blotted as the loading control. (F) Effect of the caspase inhibitor, Z-VAD-FMK, on the viability of cells treated with the indicated concentrations of TGS and MMC alone or in combination for 48 h. (T0.25, T0.5 and T0.75 represent 0.25, 0.5 and 0.75 mg/mL of TGS, respectively; M0.25, M0.5 and M0.75 represent 0.25, 0.5 and 0.75 μg/mL of MMC, respectively; MT0.25, MT0.5, and MT0.75 represent 0.25 mg/mL TGS+0.25 μg/mL MMC, 0.5 mg/mL TGS+0.5 μg/mL MMC, and 0.75 mg/mL TGS+0.75 μg/mL MMC, respectively). The date are represented as the mean±SEM (n=6). The represented cell viabilities are relative to that of each control, *P<0.05, **P<0.01 vs no Z-VAD-FMK. Student's t-test.
TGS reduces MMC-induced S-phase cell cycle arrest and DNA damage repair
As MMC could induce DNA damage, we next examined the effect of the combined treatment on cell cycle and DNA damage repair. Cellular DNA was stained with PI and detected by flow cytometry. As shown in Figures 2A and 2B, TGS significantly rescued MMC-induced S-phase arrest and led to G1-phase arrest. In addition, MMC-induced DNA damage repair occurs mainly in S-phase, as shown in both our current study and previous publications23,24, suggesting that TGS might reduce the efficiency of DNA damage repair induced by MMC in the combined treatment, and this was further confirmed by our observation of the accumulation of the DNA damage marker γ-H2AX (Figure 2C). The MMC-induced increases in the mRNA and protein levels of key genes involved in DNA damage repair, including BRCA1, BRCA2 and 53BP125,26, were also significantly inhibited by TGS (Figure 2D, Figure S3). All these data suggest that TGS could reduce MMC-induced DNA damage repair by inhibiting MMC-induced S-phase cell cycle arrest.

TGS reduces MMC-induced S-phase cell cycle arrest and DNA damage repair. (A, B) Analysis of cell cycle distribution of A549 cells treated with TGS (mg/mL) and MMC (μg/mL) alone or in combination (TGS 1, 1 mg/mL TGS; MMC 1, 1 μg/mL MMC; MT1, 1 mg/mL TGS+1 μg/mL MMC) for 24 h. The cells were stained with PI and detected by flow cytometry. The data are represented as the mean±SEM. n=3 for each group. *P<0.05, **P<0.01 vs control. #P<0.05, ##P<0.01 vs MMC 1, Student's t-test. (C) Level of γ-H2AX protein. The cells were treated with the indicated agents as in Figure 1E for 24 h, and the protein level was normalized to that of GAPDH. (D) mRNA levels of BRCA1, BRCA2 and 53BP1 in A549 cells. The cells were treated with the indicated agents for 12 h, and the mRNA levels were assessed relative to that of the control. (T0.25 and T0.5 represent 0.25 and 0.5 mg/mL of TGS, respectively; M0.5 and M1 represent 0.5 and 1 μg/mL of MMC, respectively; MT 0.5 and MT 1 represent 0.25 mg/mL TGS+0.5 μg/mL MMC and 0.5 mg/mL TGS+1 μg/mL MMC, respectively). *P<0.05, **P<0.01 vs control. Student's t-test.
The synergistic effect of TGS and MMC is dependent on the DNA repair protease Rad51
To further explore the regulatory mechanism of TGS in the repair of MMC-induced DNA damage, we next detected the mRNA levels of proteins involved in the repair of MMC-induced DNA damage and found that TGS significantly inhibited the MMC-induced upregulation of Rad51 mRNA (Figure 3A). As MEK1/2-ERK1/2-Rad51 signaling is a well-known pathway regulating the progress of DNA damage repair27, we next asked the question of whether this pathway was involved in the synergistic efficacy of TGS and MMC. As the results shown, MMC effectively induced the activation of p-MEK1/2 and p-ERK1/2 and the expression of Rad51, which was significantly blocked by TGS (Figure 3B). To validate whether the synergistic efficacy is Rad51-dependent, we silenced Rad51 with specific siRNA and treated the cells with TGS and MMC alone or in combination. The IC50 of MMC in the Rad51-silenced A549 cells decreased to approximately half of that in the control siRNA-treated cells, and Rad51 knockdown resulted in a loss of synergistic efficacy (Figure 3C). We also determined the apoptosis rates in response to TGS, MMC and their combination in A549 cells after Rad51 knockdown. The results showed an additional efficacy rather than a synergistic efficacy (Figure S4A, B, C). These data were consistent with the results of the cell viability assay. Determination of cell cycle distribution showed that TGS could still reverse the MMC-induced S-phase cell cycle arrest in the Rad51-silenced cells (Figure S4D). Investigation of the effects of ginsenoside monomers on the MEK1/2-ERK1/2-Rad51 pathway demonstrated that, among all the ginsenosides in TGS, ginsenosides Rb2 and Rh2 and diol-type ginsenosides were primarily responsible for the inhibitory effect on the pathway (Figure S5). Taken together, we concluded that the synergistic anti-cancer efficacy of the two agents TGS and MMC is dependent on the DNA repair protease Rad51.

The synergistic efficacy of TGS and MMC is dependent on the DNA repair protease Rad51. (A) Quantitative RT-PCR analysis of mRNA levels of ERCC1, FANCD2, FAN1 and Rad51 in A549 cells treated with the indicated agents (the same as that in Figure 2C) for 12 h. The data are represented as the mean±SEM. n=3 for each group. NS, not significant, *P<0.05, **P<0.01 vs control. Student's t-test. (B) Protein levels of p-MEK1/2, p-ERK1/2 and Rad51. The cells were treated with the indicated agents for 24 h (T0.5 represents 0.5 mg/mL of TGS; M0.5, M1, M2 represent 0.5, 1 and 2 μg/mL of MMC, respectively; MT 0.5 represents 0.25 mg/mL TGS+0.5 μg/mL MMC; MT 1 represents 0.5 mg/mL TGS+1 μg/mL MMC; MT2 represents 1 mg/mL TGS+2 μg/mL MMC), U0126 (10 μmol/L) was used as MEK1/2 inhibitor. GAPDH served as the loading control. (C) Effect of knockdown of Rad51 on the synergistic effect of TGS and MMC. Cellular Rad51 was silenced with 20 nmol/L Rad51-targeting siRNA, and the cells were then treated with TGS, MMC or in combination (TGS: MMC=1:1) under the same conditions for 48 h. The cell inhibition rates are relative to each control. The data are represented as the mean±SEM. n=3 for each group.
TGS inhibits the translocation of Rad51 from the cytoplasm to the nucleus
Because the translocation of Rad51 into the nucleus plays a key role in its ability to form foci and its subsequent DNA repair function28, we next studied the effect of TGS and MMC on the nuclear translocation of Rad51. The immunoblotting results showed that the level of Rad51 protein was significantly increased in the cytoplasm and decreased in the nucleus by TGS treatment (Figure 4A), suggesting an inhibition of the cytoplasm-to-nucleus transport of Rad51 by TGS. In addition, the above results were further confirmed by immunofluorescence staining for Rad51 (Figure 4B). These data suggest that TGS inhibits the translocation of Rad51 from the cytoplasm to the nucleus, which could also contribute to the synergistic efficacy of TGS with MMC.

TGS inhibits the translocation of Rad51 from the cytoplasm to the nucleus. (A) Western blot analysis of the level of Rad51 in total cell lysates, cytoplasm and nucleus of A549 cells. The cells were treated with the indicated agents (the same as that in Figure 1E, MT: MMC/TGS, TGS:MMC=1:2) for 24 h, and the levels of Rad51 in total cell lysates, cytoplasm and nucleus were detected. The purity of different cellular fractions was confirmed by Western blotting for GAPDH and histone H3, which are exclusively located in the cytoplasm and the nucleus, respectively50. (B) Immunofluorescence analysis of Rad51 translocation in cells treated with 0.5 mg/mL TGS (TGS 0.5), 1 μg/mL MMC (MMC 1) or 0.5 mg/mL TGS+1 μg/mL MMC (MT 1) for 24 h, and then Hoechst 33342 was used to stain the nuclei. The experiment was performed in triplicate, and representative images are shown. Scale bar, 20 μm.
Combined treatment of TGS with MMC inhibits NSCLC progression in vivo
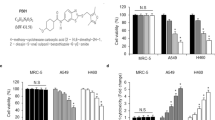
Based on the inhibitory effects in vitro, we next evaluated the effect of the combined treatment (TGS/MMC) on an established xenograft model in vivo. In agreement with the in vitro data, the combination therapy significantly inhibited tumor growth by 72.80% (TGS/MMC) compared with 10.26% (TGS) or 33.19% (MMC) as measured by the tumor volume, or by 70.74% (TGS/MMC) compared with -7.78% (TGS) or 47.64% (MMC) as measured by the tumor weight (Figure 5A, 5B, 5D and 5E). Images of all the tumors are shown in Figure 5C. Cell death in the tumors was also found to be significantly enhanced by the combination therapy when compared with TGS or MMC treatment alone by both HE staining (Figure 5F) and TUNEL immunostaining (Figure 5G). Meanwhile, Ki67 staining showed a complete absence of proliferating cells in the co-treated group (Figure 5H). All these data were consistent with the in vitro data and further confirmed the synergistic anticancer effect of TGS and MMC in vivo.

Combined treatment with TGS and MMC inhibits NSCLC progression in vivo. (A) Tumor volume (mm3) of NSCLC xenografts in mice, n=8 or 9 mice for each group and vehicle control. The data are represented as the mean±SEM. NS: not significant. *P<0.05, **P<0.01 vs control (two-tailed unpaired Student's t-test). (B) The average tumor sizes and tumor inhibition rates were calculated based on tumor volumes. (C) Images of xenografts from the control, TGS, MMC and combined TGS/ MMC treatment groups at the end of the experiment. Scale bar, 1 cm. (D) Tumor weights (mg) of each group, n.s. not significant, *P<0.05, **P<0.01 vs control. Student's t-test. (E) The average tumor sizes and tumor inhibition rates according to tumor weight. (F) HE staining of tumor tissues. Scale bar, 20 μm. (G) Immunofluorescence for TUNEL staining of tumor tissues. Scale bar, 20 μm. (H) Ki67 staining of tumor tissues. The representative images are shown. Scale bar, 50 μm.
Discussion
Our results suggest that MMC can be successfully combined with TGS to eradicate NSCLC cell lines A549 and PC-9 in vitro and to inhibit tumor growth in an established xenograft model in vivo, partly due to enhanced caspase-mediated apoptotic cell death and a compromised effect on cell cycle arrest. The combination treatment could decrease the phosphorylation of MEK1/2-ERK1/2, as well as the levels of Rad51 protein and could block the translocation of Rad51 from the cytoplasm to the nucleus. The synergistic efficacy of TGS and MMC was found to be dependent on the DNA recombination repair protease Rad51. These new findings reveal a novel strategy for the treatment of NSCLC and show that a combination of the traditional Chinese medicine TGS and the chemotherapy agent MMC may improve anticancer efficacy.
Natural compounds are indispensable sources for the development of modern drugs, and many natural products have anticancer effects; however, the targets and mechanisms of these natural compounds are unknown15,29. Ginseng is the most widely used traditional Chinese medicine in Asian countries. Although many studies on ginseng have demonstrated that several ginsenoside monomers, including Rg3, compound K and Rh2, can regulate cell death and proliferation13,30,31, suggesting a potential for ginsenosides in combating cancers, little is known about the exact role and mechanism of total ginsenosides extract in combating NSCLC.
To explore the anti-NSCLC mechanism of ginseng extract, we screened several first-line chemotherapy agents together with TGS for cytotoxicity in the NSCLC cell line A549. We found, for the first time, that TGS could significantly enhance the cytotoxicity of MMC in NSCLC A549 and PC-9 cells. The CI (combination index) calculated according to Chou22 suggests a synergistic effect of the two agents. Hoechst staining and TUNEL assay suggested that TGS increased apoptotic cell death induced by MMC. Immunoblot analysis showed that the combined treatment significantly increased the expression and activation of caspase family proteins and pro-apoptotic proteins of the Bcl-2 family. The synergistic effect of the two agents could be partly reversed by treatment with Z-VAD-FMK, a caspase inhibitor, suggesting a caspase-dependent mechanism.
TGS itself displayed slight inhibitory effects on the viability of A549 and PC-9 cells in vitro and had no significant effect in decreasing tumor size in vivo when used alone. All these data suggest that the synergistic effect of TGS and MMC is not entirely due to the direct toxicity of TGS on NSCLC A549 cells. As TGS alone could significantly induce G1-phase cell cycle arrest and could inhibit MMC-induced S-phase cell cycle arrest and efficiency of DNA damage repair, the direct regulation of the cell cycle by TGS could be a possible mechanism for its synergistic behavior with MMC against NSCLC cells.
The screen for changes in mRNA levels demonstrated a significantly rescued effect of TGS on the MMC-induced upregulation of Rad51. The MEK1/2-ERK1/2-Rad51 pathway is already known to be regulated by TGS and MMC, suggesting that it is highly likely that Rad51-mediated DNA damage repair is involved in the synergistic effect. Our observations that TGS could significantly inhibit the upregulation of phosphorylation of MEK1/2-ERK1/2 and expression of Rad51 protein induced by MMC and inhibit the translocation of Rad51 from the cytoplasm to the nucleus in NSCLC A549 cells further support this model.
RAS-RAF-MEK-ERK signaling is a crucial pathway that regulates many cellular processes, such as survival, proliferation and differentiation32,33. ERK signaling is highly activated in many tumors, which leads to drug resistance34. Many scientists have reported that a significant increase in the efficacy of chemotherapy agents and the reversal of drug resistance may be achieved by inhibiting this pathway35,36. Interstrand DNA crosslinks, adducts and double-strand breaks (DSBs) induced by MMC are lethal forms of DNA damage that need to be specifically repaired by homologous recombination6,37. Rad51 is an indispensable protein that has a key role in the homologous recombination repair pathway and is often highly expressed in tumor cells, resulting in resistance to chemotherapy agents38,39. High-levels of expression of Rad51 can be used as independent prognostic markers of survival among non-small cell lung cancer patients, and Rad51 is a potential biomarker and therapeutic target in pancreatic cancer40,41. Many drugs can enhance the anticancer efficacy of chemotherapy agents by reducing the expression of Rad5142,43; therefore, Rad51 can be used as a target for anticancer therapy44,45. Apart from the expression of Rad51, its nuclear translocation is also a key aspect of its function46. Recently, novel insights into the activity and regulation of Rad51 during homologous recombination and DNA replication have been obtained47. Chemical agonists of Rad51 are not available. Overexpression of Rad51 can contribute to chemoresistance48. Furthermore, from the results of the above studies and those in the literature, it can be predicted that the cytotoxicity of TGS, MMC and their combination in Rad51-overexpressing A549 cells would be lower than that in normal A549 cells, because of the DNA damage repair function of Rad51, and thus, TGS might not inhibit the expression of the Rad51 plasmid through the MEK1/2-ERK1/2-Rad51 pathway.
Based on the results in vitro, we examined the antitumor effect of TGS and MMC treatment alone or in combination at an appropriate dosage on an established xenograft model in vivo10,49. The combined treatment of TGS and MMC significantly increased the tumor inhibition rate and there were almost no proliferation-positive cells in the tumors compared with the treatment with TGS or MMC alone .
In summary, our results demonstrated that TGS, an anticancer adjuvant extracted from one of the most widely used traditional Chinese medicines, ginseng, could synergize with MMC to kill NSCLC A549 cells in vitro and could inhibit the growth of lung tumors in vivo. These findings demonstrated the potential use of TGS as a natural adjuvant in the clinic for the treatment of NSCLC patients with MMC.
Abbreviation
53BP1, p53 binding protein 1; BRCA1/2, breast cancer 1/2; CCK-8, cell counting kit 8; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HE staining, hematoxylin-eosin staining; MMC, mitomycin C; NSCLC, non-small cell lung carcinoma; PBS, phosphate-buffered saline; PVDF, polyvinylidene fluoride; PBST, PBS containing 0.1% Tween-20; qRT-PCR, quantitative real-time polymerase chain reaction; siRNA, small interfering RNA; SPF, specific pathogen-free; TBST, Tris-buffered saline containing 0.1% Tween-20; TGS, total ginsenosides extract.
Author contribution
Min ZHAO, Li-juan CAO, Guang-ji WANG and Hai-ping HAO participated in research design; Min ZHAO, Dan-dan WANG, Yuan CHE, Qing-ran LI and Chang SHAO conducted experiments; Yuan CHE, Meng-qiu WU and Yun WANG contributed new reagents or analytical tools; Min ZHAO, Li-juan CAO and Hai-ping HAO performed data analyses; Min ZHAO, Li-juan CAO, Hai-ping HAO and Guang-ji WANG wrote or contributed to the writing of the manuscript.
References
Torre L A, Bray F, Siegel R L, Ferlay J, Lortet-Tieulent J, Jemal A . Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108.
Brambilla E, Travis W D, Colby T V, Corrin B, Shimosato Y . The new World Health Organization classification of lung tumours. Eur Respir J 2001; 18: 1059–68.
Verges B, Walter T, Cariou B . Endocrine side effects of anti-cancer drugs: effects of anti-cancer targeted therapies on lipid and glucose metabolism. Eur J Endocrinol 2014; 170: R43–55.
Monsuez JJ, Charniot JC, Vignat N, Artigou JY . Cardiac side-effects of cancer chemotherapy. Int J Cardiol 2010; 144: 3–15.
Mayor S . Side-effects of cancer drugs are under-reported in trials. Lancet Oncol 2015; 16: e107.
Deans AJ, West SC . DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11: 467–80.
Schwartz HS, Sodergren JE, Philips FS . MItomycin c: chemical and biological studies on alkylation. Science 1963; 142: 1181–3.
Iyer VN, Szybalski W . Mitomycins and porfiromycin: chemical mechanism of activation and cross-linking of DNA. Science 1964; 145: 55–8.
Bradner WT . Mitomycin C: a clinical update. Cancer Treat Rev 2001; 27: 35–50.
Babiak A, Hetzel J, Godde F, Konig H H, Pietsch M, Hetzel M . Mitomycin C and vinorelbine for second-line chemotherapy in NSCLC--a phase II trial. Br J Cancer 2007; 96: 1052–6.
Booton R, Lorigan P, Anderson H, Baka S, Ashcroft L, Nicolson M, et al. A phase III trial of docetaxel/carboplatin versus mitomycin C/ifosfamide/cisplatin (MIC) or mitomycin C/vinblastine/cisplatin (MVP) in patients with advanced non-small-cell lung cancer: a randomised multicentre trial of the British Thoracic Oncology Group (BTOG1). Ann Oncol 2006; 17: 1111–9.
DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 2014; 64: 252–71.
Chen L, Meng Y, Sun Q, Zhang Z, Guo X, Sheng X, et al. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis 2016; 7: e2334.
Kim DG, Jung KH, Lee DG, Yoon JH, Choi KS, Kwon SW, et al. 20(S)-Ginsenoside Rg3 is a novel inhibitor of autophagy and sensitizes hepatocellular carcinoma to doxorubicin. Oncotarget 2014; 5: 4438–51.
Reddy L, Odhav B, Bhoola KD . Natural products for cancer prevention: a global perspective. Pharmacol Ther 2003; 99: 1–13.
Cragg GM, Grothaus PG, Newman DJ . Impact of natural products on developing new anti-cancer agents. Chem Rev 2009; 109: 3012–43.
Jia L, Zhao Y . Current evaluation of the millennium phytomedicine--ginseng (I): etymology, pharmacognosy, phytochemistry, market and regulations. Curr Med Chem 2009; 16: 2475–84.
Christensen LP . Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv Food Nutr Res 2009; 55: 1–99.
Yi XQ, Li T, Wang JR, Wong VK, Luo P, Wong IY, et al. Total ginsenosides increase coronary perfusion flow in isolated rat hearts through activation of PI3K/Akt-eNOS signaling. Phytomedicine 2010; 17: 1006–15.
Wong VK, Cheung SS, Li T, Jiang ZH, Wang JR, Dong H, et al. Asian ginseng extract inhibits in vitro and in vivo growth of mouse lewis lung carcinoma via modulation of ERK-p53 and NF-kappaB signaling. J Cell Biochem 2010; 111: 899–910.
Hao H, Lai L, Zheng C, Wang Q, Yu G, Zhou X, et al. Microsomal cytochrome p450-mediated metabolism of protopanaxatriol ginsenosides: metabolite profile, reaction phenotyping, and structure-metabolism relationship. Drug Metab Dispos 2010; 38: 1731–9.
Chou TC . Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–81.
Rothfuss A, Grompe M . Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol 2004; 24: 123–34.
Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD . S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 2002; 100: 2414–20.
Foulkes WD, Shuen AY . In brief: BRCA1 and BRCA2. J Pathol 2013; 230: 347–9.
Panier S, Boulton SJ . Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15: 7–18.
Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 2015; 208: 563–79.
Okimoto S, Sun J, Fukuto A, Horikoshi Y, Matsuda S, Matsuda T, et al. hCAS/CSE1L regulates RAD51 distribution and focus formation for homologous recombinational repair. Genes Cells 2015; 20: 681–94.
Hao H, Zheng X, Wang G . Insights into drug discovery from natural medicines using reverse pharmacokinetics. Trends Pharmacol Sci 2014; 35: 168–77.
Lu P, Su W, Miao Z H, Niu H R, Liu J, Hua QL . Effect and mechanism of ginsenoside Rg3 on postoperative life span of patients with non-small cell lung cancer. Chin J Integr Med 2008; 14: 33–6.
Kim MJ, Yun H, Kim DH, Kang I, Choe W, Kim SS, et al. AMP-activated protein kinase determines apoptotic sensitivity of cancer cells to ginsenoside-Rh2. J Ginseng Res 2014; 38: 16–21.
Maik-Rachline G, Seger R . The ERK cascade inhibitors: Towards overcoming resistance. Drug Resist Updat 2016; 25: 1–12.
Okumura S, Janne PA . Molecular pathways: the basis for rational combination using MEK inhibitors in KRAS-mutant cancers. Clin Cancer Res 2014; 20: 4193–9.
Samatar AA, Poulikakos PI . Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014; 13: 928–42.
Heigener DF, Gandara DR, Reck M . Targeting of MEK in lung cancer therapeutics. Lancet Respir Med 2015; 3: 319–27.
Stinchcombe TE, Johnson GL . MEK inhibition in non-small cell lung cancer. Lung Cancer 2014; 86: 121–5.
Mehta A, Haber JE . Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014; 6: a016428.
Tarsounas M, Davies AA, West SC . RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci 2004; 359: 87–93.
Richardson C . RAD51, genomic stability, and tumorigenesis. Cancer Lett 2005; 218: 127–39.
Qiao GB, Wu YL, Yang XN, Zhong WZ, Xie D, Guan XY, et al. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br J Cancer 2005; 93: 137–43.
Nagathihalli NS, Nagaraju G . RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochim Biophys Acta 2011; 1816: 209–18.
Ko JC, Chen JC, Wang TJ, Zheng HY, Chen WC, Chang PY, et al. Astaxanthin down-regulates Rad51 expression via inactivation of AKT kinase to enhance mitomycin C-induced cytotoxicity in human non-small cell lung cancer cells. Biochem Pharmacol 2016; 105: 91–100.
Ko JC, Tsai MS, Weng SH, Kuo YH, Chiu YF, Lin YW . Curcumin enhances the mitomycin C-induced cytotoxicity via downregulation of MKK1/2-ERK1/2-mediated Rad51 expression in non-small cell lung cancer cells. Toxicol Appl Pharmacol 2011; 255: 327–38.
Carvalho JF, Kanaar R . Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin Ther Targets 2014; 18: 427–58.
Ward A, Khanna KK, Wiegmans AP . Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev 2015; 41: 35–45.
Gildemeister OS, Sage JM, Knight KL . Cellular redistribution of Rad51 in response to DNA damage: novel role for Rad51C. J Biol Chem 2009; 284: 31945–52.
Godin SK, Sullivan MR, Bernstein KA . Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol 2016; 94: 407–18.
Hannay JA, Liu J, Zhu QS, Bolshakov SV, Li L, Pisters PW, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther 2007; 6: 1650–60.
Liu WY, Zhang JW, Yao XQ, Jiang C, He JC, Ni P, et al. Shenmai injection enhances the cytotoxicity of chemotherapeutic drugs against colorectal cancers via improving their subcellular distribution. Acta Pharmacol Sin 2017; 38: 264–76.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No 81325025, 81430091, 91429308, 8153000588 and 81603193) and the project for Major New Drugs Innovation and Development (No 2015ZX09501010).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Information
Supplemental Data (DOC 2272 kb)
Rights and permissions
About this article

Cite this article
Zhao, M., Wang, Dd., Che, Y. et al. Ginsenosides synergize with mitomycin C in combating human non-small cell lung cancer by repressing Rad51-mediated DNA repair. Acta Pharmacol Sin 39, 449–458 (2018). https://doi.org/10.1038/aps.2017.53
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.53
