
Abstract
Ulinastatin (UTI) is a broad-spectrum serine protease inhibitor isolated and purified from human urine with strong anti-inflammatory and cytoprotective actions, which is widely used for the treatment of various diseases, such as pancreatitis and sepsis. Although the therapeutic effects of UTI are reported to be associated with a variety of mechanisms, the signaling pathways mediating the anti-inflammatory action of UTI remain to be elucidated. In the present study we carried out a systematic study on the anti-inflammatory and anti-oxidative mechanisms of UTI and their relationships in LPS-treated RAW264.7 cells. Pretreatment with UTI (1000 and 5000 U/mL) dose-dependently decreased the mRNA levels of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α, iNOS) and upregulated anti-inflammatory cytokines (IL-10 and TGF-β1) in LPS-treated RAW264.7 cells. UTI pretreatment significantly inhibited the nuclear translocation of NF-κB by preventing the degradation of IκB-α. UTI pretreatment only markedly inhibited the phosphorylation of JNK at Thr183, but it did not affect the phosphorylation of JNK at Tyr185, ERK-1/2 and p38 MAPK; JNK was found to function upstream of the IκB-α/NF-κB signaling pathway. Furthermore, UTI pretreatment significantly suppressed LPS-induced ROS production by activating PI3K/Akt pathways and the nuclear translocation of Nrf2 via promotion of p62-associated Keap1 degradation. However, JNK was not involved in mediating the anti-oxidative stress effects of UTI. In summary, this study shows that UTI exerts both anti-inflammatory and anti-oxidative effects by targeting the JNK/NF-κB and PI3K/Akt/Nrf2 pathways.
Similar content being viewed by others


Introduction
Ulinastatin (UTI) is a broad-spectrum serine protease inhibitor that is isolated and purified from human urine, with strong anti-inflammatory and cytoprotective actions in various cell and animal models1. UTI inhibits trypsin and is commonly used in the treatment of pancreatitis and sepsis2,3. In addition, UTI stabilizes lysosomal membranes and inhibits lysosomal enzyme release and myocardial depressant factor production1. UTI also inhibits neutrophil activation and transendothelial migration, reduces inflammatory cell infiltration and down-regulates inflammatory cytokines and is currently used for the treatment of acute circulatory failure4. Early administration of UTI has been demonstrated to inhibit neutrophil protease release and excessive inflammatory responses, and reduce the level of oxygen free radicals and the consumption of superoxide dismutase5. Although the anticoagulant and organ-protective effects of UTI may be partially explained by inhibition of protease activities, the anti-inflammatory action of UTI cannot be fully explained by protease inhibition. Mechanisms mediating the anti-inflammatory action of UTI remain to be elucidated.
Anti-inflammatory mechanisms work through different pathways. Increasing evidence suggests that nuclear factor-kappa B (NF-κB) is a central transcriptional factor in the regulation of inflammatory factors6, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, nitric oxide (NO) and others7,8,9. Among these mediators, one of great concern is NO produced by inducible NO synthase (iNOS)10,11. Anti-inflammatory cytokines are a series of molecules that reduce the production and activity of pro-inflammatory cytokines, and regulate the inflammatory response, such as IL-10 and transforming growth factor (TGF)-β112,13. In addition, mitogen-activated protein kinases (MAPKs) are classic inflammation-related signals, and include extracellular signal-related kinase (ERK)-1/2, p38, and c-Jun NH2-terminal kinase (JNK). Among these, JNK and p38 are mainly involved in the expression of pro-inflammatory mediators14. Therefore, MAPKs might play important roles in the anti-inflammatory mechanisms of UTI.
Oxygen metabolism also has an important role in the pathogenesis of inflammation. Increasing evidence suggests that reactive oxygen species (ROS) may be the critical molecular mechanism underlying various inflammatory-associated diseases. As previously reported, an increase in the production of ROS has been implicated in inflammatory arthritis15,16. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator in oxidative stress and the inflammatory response. Nrf2 dissociates from Kelch-like ECH-associated protein1 (Keap1), translocates to the nucleus, binds to the antioxidant response element (ARE), and triggers the expression of ARE-regulated genes, including heme oxygenase-1 (HO-1), which is an important inducible antioxidant enzyme17,18. Therefore, UTI might perform its role in anti-inflammation through the inhibition of ROS overproduction, by activating the Nrf2 pathway in LPS-stimulated RAW264.7 cells.
In this study, we examined the effects of UTI on the expression of inflammatory cytokines and mediators, and integrated the molecular mechanisms of anti-inflammatory and anti-oxidative effects in LPS-stimulated RAW264.7 cells to demonstrate the interrelationship of these signaling pathways.
Materials and methods
Reagents
UTI was from Techpool Bio-pharma Co, Ltd (Guangzhou, China). LPS (Escherichia coli 055:B5) was from Sigma-Aldrich (St Louis, MO, USA). DMEM, FBS, and TRIzol reagent were purchased from Gibco BRL (Gaithersburg, MD, USA). Nuclear Protein Extraction kit was from Cayman Chemical Company (MI, USA). Primers for quantitative real-time polymerase chain reaction (qRT-PCR) were synthesized by Sangon Biotech (Guangzhou, China). SP600125 and wortmannin were from LC Laboratories (Woburn, MA, USA).
Antibodies to NF-κB p65 (sc-109), β-actin (sc-5274) and IκB-α (sc-1643) were from Santa Cruz (Santa Cruz, CA, USA); antibodies to p-PI3K (4228), PI3K (4249), p-Akt (2965), Akt (4691), p-MKK4 (4514), MKK4 (9152), MKK7 (4172), p-JNK (4668), JNK (9258), p-p38 (4511), p38 (8690), p-ERK (4370), ERK (4695), and Nrf2 (12721) were from Cell Signaling Technology (Danvers, MA, USA); antibody to p-MKK7 (abs137245) was from Absin (Absin Bioscience, Shanghai, China); antibody to p62 (PM045) was from MBL International (Woburn, MA, USA); antibodies to Keap1 (10503-2-AP), and HO-1 (10701-AP) were from Proteintech; antibody to iNOS (ab178945) was from Abcam (Cambridge, UK). Secondary antibodies conjugated with Alexa Fluor-488 were from Invitrogen.
Cell culture
RAW264.7 cells, a mouse macrophage cell line, were purchased from the Cell Bank of the Shanghai Institute of Cell Biology and Biochemistry, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in high glucose DMEM, supplemented with 10% (v / v) FBS, 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in a humidified air atmosphere with 5% (v/v) CO2. UTI was dissolved in PBS to a concentration of 105 U/mL and further diluted in cell culture media.
Total RNA extraction and qRT-PCR analysis
RAW264.7 cells (1.5×105 cells in a 12-well plate) were pretreated with UTI (1000 or 5000 U/mL) for 3 h, then LPS (1 μg/mL) was added for 6 h or 12 h at 37 °C in a humidified air atmosphere with 5% (v / v) CO2. Total RNA was extracted with TRIzol Reagent and 4 μg total RNA was reverse-transcribed using random hexamers at 37°C with the M-MLV reverse transcriptase (Promega, Madison, USA). qRT-PCR was performed using SYBR Green (GeneCopoeia, USA) in a RT-PCR machine (QuantStudio 6 Flex System, Applied Biosystems, Foster City, CA, USA). The relative levels of gene expression were calculated as ΔCt=Ct (gene)-Ct (reference). The 2-ΔΔCt method was used to calculate the fold-change of gene expression. The sequences of primers used are listed in Table 1.
Western blot analysis
Cell lysates were prepared using RIPA buffer with a protease and phosphatase inhibitor cocktail (Thermo Scientific, USA). Equal amounts of protein (25 μg/lane) were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The PVDF membranes were then blocked for 1.5 h at room temperature with 5% (w/v) non-fat dried milk in TBST. Specific proteins were then detected using appropriate primary antibodies and horseradish peroxidase-conjugated secondary antibodies, followed by enhanced chemiluminescence development (Millipore). Images were digitally acquired with ImageQuant LAS 4000mini (GE, Uppsala, Sweden), and companion software.
Immunofluorescence staining
RAW264.7 cells were preincubated in 96-well plates and pretreated with UTI (1000, 5000 U/mL) for 3 h, then LPS (1 μg/mL) was added for 15 min at 37 °C in a humidified atmosphere with 5% (v / v) CO2. Cells were washed two times with PBS, permeabilized with 0.1% Triton X-100 for 15 min, followed by another wash in PBS and blocking in PBS containing 5% bovine serum albumin (BSA) for 1 h. Primary and secondary antibodies in PBS containing 5% BSA were applied overnight at 4 °C and 1 h at room temperature, respectively. Fluorescence images were taken using an EVOS FL Auto (Life Technologies, Bothell, WA, USA).
Measurement of ROS production
ROS production was detected with 2',7'-dichlorodihydro-fluorescein diacetate (DCFH-DA) (Beyotime). RAW264.7 cells, cultured in 96-well plates for 24 h before the experiment, were pretreated with the indicated concentrations of compounds and then stimulated with LPS (1 μg/mL) for 12 h to induce ROS production. Cells were washed twice with PBS and then incubated with DCFH-DA probe (5 μmol/L) for 20 min at 37 °C. The signal of DCF (the oxidation product of DCFH-DA) was then immediately observed using the Array Scan VII (Thermo) at an excitation wavelength of 488 nm and emission wavelength of 525 nm. The average fluorescence intensity from randomly selected fields (10 for each group) was measured using the built-in image analysis software.
Statistical analysis
All experiments were performed in at least triplicate and data were presented as the mean±SD. Differences between multiple groups were analyzed using one-way ANOVA, followed by a Tukey-Kramer or Dunnett's multi-comparison test for post hoc comparison. P<0.05 was considered statistically significant.
Results
UTI decreases pro-inflammatory cytokines and increases anti-inflammatory cytokines in LPS-treated RAW264.7 cells
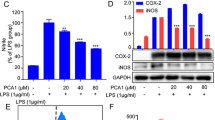
To investigate the anti-inflammatory effect of UTI on LPS-treated RAW264.7 cells, the cells were treated with UTI for 1 h prior to LPS. qRT-PCR was employed to measure mRNA expression of IL-6, IL-1β, TNF-α and iNOS, which are considered classic pro-inflammatory cytokines with a main function of recruiting additional immune cells to inflammatory sites. As shown in Figure 1, overproduction of IL-6 and IL-1β were typically observed in RAW264.7 cells after stimulation with LPS for 6 h, while excessive production of TNF-α and iNOS was detected at 12 h. As expected, pretreatment with UTI resulted in a significant decrease in IL-1β, IL-6, TNF-α and iNOS levels (Figure 1A–D). It is well-known that the generation of pro-inflammatory mediators such as NO in LPS-stimulated RAW264.7 cells is catalyzed by iNOS, and further analyses of protein expression by Western blot confirmed that the level of iNOS was consistent with its mRNA expression pattern (Figure 1E–F). Anti-inflammatory cytokines control LPS-induced release of pro-inflammatory cytokines, so we further studied changes in IL-10 and TGF-β1 production. RAW264.7 cells were exposed to LPS for 12 h after pretreatment with UTI. Following this treatment, we found that high concentrations of UTI significantly increased IL-10 production compared to LPS-treated groups (Figure 1G), and effectively reversed LPS-mediated TGF-β1 reduction (Figure 1H). These results indicated that UTI could markedly inhibit the inflammatory response in LPS-stimulated RAW264.7 cells.

UTI decreases pro-inflammatory cytokines and increases anti-inflammatory cytokines in LPS-treated RAW264.7 cells. Cells were pretreated with UTI (1000 or 5000 U/mL) for 3 h and then stimulated with LPS (1 μg/mL). After 6 h stimulation, the mRNA levels of IL-1β (A) and IL-6 (B) were measured by qRT-PCR. After 12 h stimulation, the mRNA levels of TNF-α (C), iNOS (D), IL-10 (G) and TGF-β1 (H) were analyzed by qRT-PCR, and the protein expression of iNOS (E–F) was detected by Western blot. The data are presented as the mean±SD of three independent experiments. * P<0.05, ** P<0.01. NS, not significant.
UTI blocks LPS-triggered activation of the IκB-α/NF-κB signaling pathway
Activation of NF-κB induces the production of pro-inflammatory cytokines, inhibits anti-inflammatory cytokines, and ultimately leads to inflammatory responses. To determine whether UTI blocks LPS-induced NF-κB activation, alterations in the level of IκB-α and nuclear translocation of NF-κB p65 were compared by Western blot and immunofluorescence staining, respectively, between cells treated with LPS-alone or LPS plus UTI for 15 min. As shown in Figure 2A, LPS stimulation markedly accelerated the degradation of IκB-α, and promoted the translocation of cytosolic NF-κB p65 into the nucleus. However, UTI effectively inhibited LPS-induced IκB-α down-regulation, and blocked the translocation of cytosolic NF-κB p65 into the nucleus. Immunofluorescence staining of NF-κB also revealed that UTI suppressed NF-κB p65 nuclear translocation in LPS-stimulated RAW264.7 cells (Figure 2B). To confirm whether UTI has a long-acting inhibitory effect on LPS-induced activation of the IκB-α/NF-κB pathway, long-term treatment of UTI was performed. As shown in Figure 2C, UTI could make IκB-α free for LPS-induced degradation, while leaving the total amount of NF-κB unchanged. Thus, UTI could block LPS-triggered activation of the IκB-α/NF-κB signaling pathway in RAW264.7 cells

UTI suppresses the IκB-α/NF-κB signaling pathway in RAW264.7 cells. Cells were pretreated with or without UTI (1000, 5000 U/mL) for 3 h, followed by incubation with LPS (1 μg/mL) for 15 min (A, B) or 12 h (C). (A) Nuclear fractions and cytoplasmic fractions were extracted and subjected to immunoblot analysis using an anti-p65 antibody and an anti-IκB-α antibody. Lamin B1 and β-actin expression were measured to confirm equal protein amounts in nuclear extracts and cytoplasmic fractions. (B) The nuclear translocation of NF-κB p65 was visualized by immunofluorescence. The nuclei were counterstained with DAPI (blue). Scale bar=200 μm. (C) The total protein levels of NF-κB p65 and IκB-α were measured by Western blot. The data are presented as the mean±SD of three independent experiments. * P<0.05, ** P<0.01. NS, not significant.
Inhibition of Thr183p-JNK by UTI was involved in NF-κB signaling pathway
The MAPKs play important roles in regulating cell growth and differentiation, as well as in the control of cellular responses to cytokines and various stresses. Moreover, they are important for activating NF-κB19. We next studied whether UTI inhibition of NF-κB activation was mediated by the inactivation of MAPKs. As shown in Figure 3A, LPS treatment markedly induced the transient phosphorylation of JNK1/2, ERK1/2 and p38 within 15 min. Western blot results showed that LPS-induced JNK phosphorylation at Thr183 was noticeably inhibited by UTI. However, UTI had no significant effect on JNK phosphorylation at Tyr185, or the phosphorylation of ERK1/2 and p38. Similarly, a 1 h pretreatment with SP600125 (a JNK inhibitor) inhibited LPS-induced IκB-α degradation and nuclear translocation of NF-κB p65 after 15 min of LPS treatment (Figure 3B). Immunofluorescence staining of NF-κB also revealed that SP600125 could reverse NF-κB p65 nuclear translocation in LPS-stimulated RAW264.7 cells (Figure 3C). Further analyses of iNOS by Western blot confirmed that SP600125 had an anti-inflammatory effect as well (Figure 3D). Intriguingly, the inhibitory effects of UTI and SP600125 on JNK were not exactly the same. Our study first found that UTI had a site-specific effect towards the dephosphorylation of JNK, mainly to dephosphorylate Thr183 (Figure 3A), whereas SP600125 could non-selectively dephosphorylate both Thr183 and Tyr185 residues on JNK (Figure 3B). Further analysis of the phosphorylation level of MKK4 and MKK7 by Western blot indicated that UTI specifically down-regulated JNK phosphorylation at Thr183, mainly through the regulation of p-MKK7, which is upstream of Thr residues, and also revealed that UTI had no significant effect on p-MKK4, the upstream kinase of Tyr residues (Figure 3E). These results suggested that UTI suppressed LPS-induced NF-κB activation partially by inhibiting LPS-induced Thr183p-JNK signaling pathways and thus exerted a potent anti-inflammatory effect.

UTI inhibits Thr183p-JNK. (A) The effects of UTI on the phosphorylation of JNK, ERK1/2 or p38 in LPS-treated RAW264.7 cells. Cells were incubated for 3 h with or without UTI (1000 or 5000 U/mL), then the cells were incubated with LPS (1 μg/mL) for 15 min, and aliquots of cell lysates containing equal amounts of protein were subjected to immunoblotting. (B–C) Cells were pretreated with or without UTI (5000 U/mL) for 3 h and SP600125 (SP, 10 μmol/L) for 1 h before co-incubation with LPS for 15 min. Nuclear fractions and cytoplasmic fractions were extracted and subjected to immunoblot analysis using an anti-p65 antibody and an anti-IκB-α antibody (B). The nuclear translocation of NF-κB p65 was observed by immunofluorescence. The nuclei were counterstained with DAPI (C). Scale bar = 200 μm. (D) After co-incubation with LPS for 12 h, the total protein levels of iNOS were measured by Western blot. (E) Cells were treated as (A). After co-incubation with LPS for 15 min, the total protein levels of p-MKK4 and p-MKK7 were measured by Western blot. The data are presented as the mean±SD of three independent experiments. * P<0.05, ** P<0.01. NS, not significant.
UTI protects against LPS-induced oxidative injury through the Nrf2/Keap1 pathway
LPS-induced excessive ROS are considered to be a key factor in promoting the development of inflammation, since they can further induce the expression of cytokines in macrophages. To assess the amount of ROS generated by LPS, as well as the ability of UTI to relieve this effect, we tested the production of ROS with the molecular probe DCFH-DA. The results showed that UTI noticeably reduced LPS-induced ROS generation (Figure 4A). Nrf2 is suggested to protect cells from excessive ROS during the inflammatory response. Nrf2 also directly regulates the expression of inflammation-associated genes, such as IL-620. Here, we further analyzed the effect of UTI on the modulation of nuclear Nrf2 protein in LPS-induced inflammation in RAW264.7 cells. As shown in Figure 4B, LPS alone had little impact on the nuclear translocation of Nrf2, but UTI significantly promoted the translocation of cytosolic Nrf2 into the nucleus. Interestingly, we observed that Keap1 translocation into the cytoplasm increased when more UTI was added. Based on the above results, we inferred that UTI played an anti-inflammatory effect that might be partially due to the regulation of the Nrf-2/HO-1 signaling pathway. We then examined the protein expression levels of Nrf2 and HO-1 and found that UTI treatment apparently increased the level of Nrf2 and HO-1 (Figure 4C). Additionally, Keap1, the suppressor of Nrf2, was significantly degraded in the UTI group. It has been reported that p62 modulates the Nrf2-Keap1 signaling pathway by directly interacting with Keap1 to promote the autophagic degradation of Keap121. Hence, we determined whether the Nrf2 activation mediated by UTI was a result of p62 accumulation. As expected, UTI significantly induced p62 accumulation (Figure 4C). Importantly, UTI itself did not activate the Nrf-2/HO-1 pathway (Figure 4D). These data suggested that UTI protected against LPS-induced ROS through activation of Nrf2 by promoting p62-associated Keap1 degradation.

UTI protects against LPS-induced ROS stress. RAW264.7 cells were pretreated with or without UTI (1000 or 5000 U/mL) for 3 h, followed by exposure to LPS (1 μg/mL). (A) After 12 h, the products of ROS were detected using DCFH-DA, and the average fluorescence intensity from randomly selected fields (10 for each group) was measured. Scale bar=400 μm. (B) After 15 min, nuclear fractions and cytoplasmic fractions were extracted and subjected to Western blotting. (C) After 12 h, expression of Nrf2 and Keap1, as well as p62 and HO-1 was detected by Western blotting. (D) Cells were pre-incubated for 3 h with UTI (5000 U/mL) and were then treated with or without LPS for 12 h. The data are presented as the mean±SD of three independent experiments. * P<0.05, ** P<0.01. NS, not significant.
To better understand the mechanism of UTI in preventing oxidation pathways, the PI3K/Akt pathway was investigated, which is an important upstream regulator of Nrf-2/HO-1. Our results showed that UTI significantly enhanced the phosphorylation of PI3K and Akt (Figure 5A). However, UTI itself did not have such effects (Figure 5B). Additionally, pretreatment with a PI3K/Akt inhibitor, wortmannin, could completely suppress the activation of PI3K and Akt phosphorylation (Figure 5C). Moreover, wortmannin also abolished the UTI-mediated expression of Nrf2, Keap1 and HO-1 after 12 h of LPS treatment (Figure 5D). Thus, the protective role of UTI with regards to anti-oxidation is mainly through PI3K/AKT-mediated activation of the Nrf2/Keap1 pathway.

UTI activates the PI3K/Akt pathway in an LPS model. (A) RAW264.7 cells were pretreated with or without UTI (1000 or 5000 U/mL) for 3 h before exposure to LPS (1 μg/mL) for 15 min. Immunoblotting and densitometric analysis were performed. (B) Cells were pretreated with UTI (5000 U/mL) for 3 h, and then were stimulated with or without LPS for 15 min. (C) Cells were pretreated with or without UTI (5000 U/mL) for 3 h and wortmannin (10 μmol/L) for 1 h before LPS treatment for 15 min. Expression levels of p-PI3K, PI3K, p-Akt and Akt were detected by Western blot. (D) Cells were treated and (C), after treatment with LPS for 12 h, expression levels of Nrf2, Keap1 and HO-1 were detected. The data are presented as the mean ± SD of three independent experiments. *P<0.05; **P<0.01; NS, not significant.
Activation of Nrf2 by UTI does not depend on JNK
In different inflammatory models, the activation of the Nrf2 pathway can be affected by multiple signal kinases. Our studies have shown that UTI can activate the Nrf2 pathway, which is partially regulated by PI3K/Akt. We further investigated whether JNK was involved in Nrf2 activation in LPS-induced inflammatory models. The results showed that SP600125 had no effects on PI3K and Akt phosphorylation after 15 min of LPS treatment (Figure 6A). Additionally, the inhibition of JNK did not promote the translocation of Nrf2 or Keap1 (Figure 6B). Moreover, pretreatment with SP600125 did not alter the expression of Nrf2, p62, Keap1 and HO-1 after co-incubation with LPS for 12 h (Figure 6C). DCFH-DA staining of ROS also revealed that SP600125 did not reduce LPS-induced ROS (Figure 6D, 7). The above results indicated that PI3K and Nrf2 signals were not downstream of JNK and that JNK did not participate in the anti-oxidative effects of UTI.

The activation effect of UTI on the Nrf2 pathway is JNK-independent. (A–D) RAW264.7 cells were pretreated with or without UTI (5000 U/mL) for 3 h, and with or without SP600125 (10 μmol/L) for 1 h before co-incubating with LPS. After 15 min, (A) immunoblotting and densitometric analysis showed p-PI3K, PI3K, p-Akt and Akt protein levels in RAW264.7 cells. (B) Nuclear fractions and cytoplasmic fractions were extracted and subjected to Western blotting. (C) After 12 h, expression of Nrf2 and Keap1, as well as p62 and HO-1 was detected. (D) Cells were treated and (C), production of ROS was detected using DCFH-DA. Scale bar=400 μm. The data are presented as the mean±SD of three independent experiments. * P<0.05; ** P<0.01. NS, not significant.

Scheme summarizing the protective effects of UTI on LPS-induced RAW264.7 cells. Ulinastatin (UTI) treatment effectively suppressed LPS-induced inflammatory injury. This is largely dependent on the suppression of LPS-activated JNK phosphorylation at Thr183, thereby inhibiting IκBα down-regulation and the NF-κB signaling pathway. In addition, UTI performed its role in anti-inflammation through ROS suppression, which mainly relies on the upregulation of the Nrf2-mediated antioxidation pathway by promoting p62-associated Keap1 degradation and the PI3K/Akt pathway. Meanwhile, the JNK signaling pathway is not involved in mediating the Nrf2 pathway.
Discussion
Here, we demonstrated that UTI treatment significantly attenuated the inflammatory response in LPS-stimulated RAW264.7 cells. This influence was partially mediated through the inhibition of Thr183p-JNK, which is upstream of NF-κB. Furthermore, UTI protected against LPS-induced oxidative stress through the activation of Nrf-2/HO-1 by promoting p62-associated Keap1 degradation. Moreover, we found that JNK did not participate in the process of UTI activation of the Nrf-2/HO-1 signaling pathway.
Macrophages are the primary responders to inflammation, and induce immune responses to endogenous and exogenous stimuli, playing a central role in host defense and immune regulation. To objectively describe the anti-inflammatory molecular mechanisms of UTI in LPS-stimulated RAW264.7 cells, we chose different incubation times with LPS. Short-term treatment for 15 min was used for transient tests of protein phosphorylation and nuclear translocation, and long-term treatment for 12 h was used for tests protein expression levels. MAPKs are important kinases in macrophage anti-inflammatory signaling, while JNK is known as a stress-activated protein kinase and plays key roles in inflammation, cellular stress, and apoptosis14,22. Our results showed that LPS treatment potentiated the phosphorylation of JNK, p38 and ERK, which is consistent with studies showing that LPS affected MAPK signaling pathways in other cell types. The results from our present study clearly showed that the JNK signaling pathway was involved in mediating the anti-inflammatory effects of UTI, which was also confirmed by use of the JNK inhibitor, SP600125. However, independent from the role of SP600125, UTI specifically dephosphorylated JNK at Thr183 and is mainly opposed by MKK7, which is primarily activated in response to cytokines. Phosphorylation of Thr residues by MKK7 has been shown to be a prerequisite for JNK activation, at least for pro-inflammatory cytokines22,23. Moreover, we have tested the expression levels of p-MKK7 and p-MKK4, which are the upstream signaling molecules for the phosphorylation of Thr and Tyr sites that control JNK, respectively. Our data demonstrated, for the first time, that UTI has a site-specific effect, for the dephosphorylation of JNK at Thr183, mainly through the regulation of p-MKK7 instead of p-MKK4.
In unstimulated cells, NF-κB is normally present in the cytosol, along with the inhibitory IκB protein family. Upon stimulation with LPS, the IκB-α protein is phosphorylated and degraded, promoting NF-κB to translocate into the nucleus and to initiate transcription of inflammatory genes. We have shown that UTI could block LPS-triggered activation of the IκB-α/NF-κB signaling pathway and of JNK in RAW264.7 cells, which is consistent with other anti-inflammatory functions of UTI. It is noteworthy that recent studies have indicated that MAPKs may participate in the activation of the NF-κB pathway24,25, while other studies suggest that they may not26,27. To elucidate the relationship of the two pathways, we analyzed phosphorylation sites of JNK to compare the differences between UTI and JNK inhibitors. Although both treatments support the hypothesis that JNK is upstream of NF-κB, the dephosphorylation of JNK at Thr183, but not at Tyr185, was effectively inhibited by UTI. Therefore, we concluded that the anti-inflammatory effect of UTI is, at least in part, due to inactivation of the Thr183p-JNK/NF-κB signaling pathway.
Increasing evidence has shown that activated macrophages enhance oxygen depletion, which causes the overproduction of ROS15. HO-1, an anti-oxidative enzyme regulated by Nrf2 activation, is essential for redox homeostasis by preventing the generation of ROS. Further studies have shown that anti-oxidation signals, via the Nrf-2/HO-1 pathway, counteract inflammatory responses in various macrophage subtypes17,18 and inflammatory associated diseases28. There was a large amount of ROS in the LPS-induced inflammatory cell model, whereas pre-incubation with UTI significantly promotes the nuclear translocation of Nrf2, followed by a reduction in the total amount of ROS. Hence, these results suggest that UTI protects against LPS-induced ROS stress through activation of the Nrf2/HO-1 signaling pathway.
Nrf2 dissociates from Keap1, translocates to the nucleus, binds to the ARE, and activates transcription aimed at cytoprotective and anti-oxidative factors, including HO-1. Therefore, Keap1 degradation is important for the maintenance of cellular redox homeostasis under oxidative stress. It is tempting to speculate that down-regulation of the Keap1 protein level by UTI was mediated by the induction of Keap1 degradation. As expected, UTI treatment significantly accelerates the degradation of Keap1, leading to Nrf2 translocation. Substantial evidence has demonstrated that p62 also activates the Nrf2 pathway, since the formation of p62-Keap1-LC3 protein complexes results in autophagic degradation of Keap129,30. Notably, UTI significantly stimulated p62 expression, suggesting that UTI-induced Keap1 degradation might be related to autophagic degradation. Different pathways may activate Nrf2 in different disease models, including PI3K/Akt, AMPK and MAPK pathways 31,32,33. Crosstalk between the PI3K/Akt and Nrf2 signaling pathways is capable of protecting cells against inflammatory and oxidative damage. We confirmed that in line with the pattern of Nrf2 activation, UTI augments the phosphorylation of PI3K/Akt as well, which means that both Nrf2/HO-1 and upstream PI3K/Akt pathways are the main pathways for the anti-oxidative function of UTI.
It has been reported that MAPKs are important kinases in HUVEC redox signaling34. Low-doses of ROS could serve as signaling molecules to activate various signaling pathways, including MAPK pathways, while phosphorylated MAPKs could, in turn, produce more ROS35. UTI attenuates LPS-induced oxidative damage to human endothelial cells by suppressing the JNK/c-Jun signaling pathway36. It is apparent that anti-inflammatory and anti-oxidative signaling pathways are complex and diverse. It is still unknown whether JNK is responsible for UTI-mediated anti-oxidation responses in LPS-stimulated RAW264.7 cells. We found that UTI, but not SP600125 alone, could activate the Nrf-2/HO-1 signaling pathway. In addition, the JNK signaling pathway was not involved in mediating the anti-oxidative stress effects of UTI, suggesting that MAPKs are not always involved in the Nrf2 signaling pathway.
In this study, we demonstrated that UTI has a stronger anti-inflammatory and anti-oxidative function in LPS-treated RAW264.7 cells. Since inflammation and oxidative stress are two biological events that are relatively independent but closely linked in many pathological processes, our findings demonstrated that the protective effects of UTI on LPS-induced RAW264.7 cells can be attributed to the activation of the PI3K/Akt-Nrf2 axis and the inhibition of the Thr183p-JNK/NF-κB axis. UTI effectively suppressed LPS-induced inflammatory damage, which is largely associated with the suppression of Thr183p-JNK, thereby inhibiting IκB-α down-regulation and the NF-κB signaling pathway. In addition, UTI performed its anti-inflammatory role through ROS suppression, which is mainly due to upregulation of the Nrf2-mediated anti-oxidation pathway by promoting p62-associated Keap1 degradation. Meanwhile, the JNK signaling pathway is not involved in mediating anti-oxidative stress by UTI. Thus, our findings may provide new insights into the pathways involved in UTI's effects on the treatment of inflammatory diseases.
Author contribution
Min LI, Pei-qing LIU, and Si-tong LI conceived and designed the experiments; Si-tong LI, Qi DAI, Shu-xian ZHANG and Shu-hong LU performed the experiments; Si-tong LI, Min LI, Qiu-qiong YU, Ya-jun LIU and Fei TAN analyzed the data and wrote the paper; Pei-qing LIU, Chuan WANG, Jian-wen CHEN, He-qing HUANG discussed and edited the manuscript; Min LI, Pei-qing LIU, Qiu-qiong YU and Fei TAN contributed reagents. All authors contributed to revising the paper.
References
Inoue K, Takano H. Urinary trypsin inhibitor as a therapeutic option for endotoxin-related inflammatory disorders. Expert Opin Invest Drugs 2010; 19: 513–20.
Linder A, Russell JA. An exciting candidate therapy for sepsis: ulinastatin, a urinary protease inhibitor. Intensive Care Med 2014; 40: 1164–7.
Pan Y, Fang H, Lu F, Pan M, Chen F, Xiong P, et al. Ulinastatin ameliorates tissue damage of severe acute pancreatitis through modulating regulatory T cells. J Inflammation 2017; 14: 7.
Zhang H, Tan C, Wang X, Kang D, Chen Y, Xiong J, et al. Preventive effects of ulinastatin on complications related to pancreaticoduodenectomy: a Consort-prospective, randomized, double-blind, placebo-controlled trial. Medicine 2016; 95: e3731.
Song D, Song G, Niu Y, Song W, Wang J, Yu L, et al. Ulinastatin activates haem oxygenase 1 antioxidant pathway and attenuates allergic inflammation. Br J Pharmacol 2014; 171: 4399–412.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 2008; 132: 344–62.
Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol 2017; 18: 861–9.
Pinho-Ribeiro FA, Zarpelon AC, Mizokami SS, Borghi SM, Bordignon J, Silva RL, et al. The citrus flavonone naringenin reduces lipopolysaccharide-induced inflammatory pain and leukocyte recruitment by inhibiting NF-kappaB activation. J Nutrit Biochem 2016; 33: 8–14.
Zucoloto AZ, Manchope MF, Staurengo-Ferrari L, Pinho-Ribeiro FA, Zarpelon AC, Saraiva AL, et al. Probucol attenuates lipopolysaccharide-induced leukocyte recruitment and inflammatory hyperalgesia: effect on NF-small ka, CyrillicB activation and cytokine production. Eur J Pharmacol 2017; 809: 52–63.
Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol 2002; 2: 787–95.
Yamanishi R, Yoshigai E, Okuyama T, Mori M, Murase H, Machida T, et al. The anti-inflammatory effects of flavanol-rich lychee fruit extract in rat hepatocytes. PLoS One 2014; 9: e93818.
Hedrich CM. Shaping the spectrum–from autoinflammation to autoimmunity. Clin Immunol 2016; 165: 21–8.
Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997; 112: 321S.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–83.
Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radical Biol Med 2000; 28: 1317–27.
Xue EX, Lin JP, Zhang Y, Sheng SR, Liu HX, Zhou YL, et al. Pterostilbene inhibits inflammation and ROS production in chondrocytes by activating Nrf2 pathway. Oncotarget 2017; 8: 41988–2000.
Liu XH, Wang XL, Xin H, Wu D, Xin XM, Miao L, et al. Induction of heme oxygenase-1 by sodium 9-hydroxyltanshinone IIA sulfonate derivative contributes to inhibit LPS-mediated inflammatory response in Macrophages. Cell Physiol Biochem 2015; 36: 1316–30.
Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radical Biol Med 2015; 88: 108–46.
Ru JY, Xu HD, Shi D, Pan JB, Pan XJ, Wang YF. Blockade of NF-kappaB and MAPK pathways by ulinastatin attenuates wear particle-stimulated osteoclast differentiation in vitro and in vivo. Biosci Rep 2016; 36.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 2016; 7: 11624.
Yin S, Cao W. Toll-like receptor signaling induces Nrf2 pathway activation through p62-triggered Keap1 degradation. Mol Cell Biol 2015; 35: 2673–83.
Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997; 112: 321s–29s.
Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Develop 2001; 15: 1419–26.
Wang W, Guan WJ, Huang RQ, Xie YQ, Zheng JP, Zhu SX, et al. Carbocisteine attenuates TNF-alpha-induced inflammation in human alveolar epithelial cells in vitro through suppressing NF-kappaB and ERK1/2 MAPK signaling pathways. Acta Pharmacol Sin 2016; 37: 629–36.
Meng Z, Yan C, Deng Q, Gao DF, Niu XL. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-kappaB pathways. Acta Pharmacol Sin 2013; 34: 901–11.
Chen HB, Luo CD, Liang JL, Zhang ZB, Lin GS, Wu JZ, et al. Anti-inflammatory activity of coptisine free base in mice through inhibition of NF-kappaB and MAPK signaling pathways. Eur J Pharmacol 2017.
Wu J, Zhang H, Hu B, Yang L, Wang P, Wang F, et al. Coptisine from Coptis chinensis inhibits production of inflammatory mediators in lipopolysaccharide-stimulated RAW 264. 7 murine macrophage cells. Eur J Pharmacol 2016; 780: 106–14.
Li Z, Ma QQ, Yan Y, Xu FD, Zhang XY, Zhou WQ, et al. Edaravone attenuates hippocampal damage in an infant mouse model of pneumococcal meningitis by reducing HMGB1 and iNOS expression via the Nrf2/HO-1 pathway. Acta Pharmacol Sin 2016; 37: 1298–306.
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30: 3275–85.
Su X, Huang Q, Chen J, Wang M, Pan H, Wang R, et al. Calycosin suppresses expression of pro-inflammatory cytokines via the activation of p62/Nrf2-linked heme oxygenase 1 in rheumatoid arthritis synovial fibroblasts. Pharmacol Res 2016; 113: 695–704.
Bai Y, Wang X, Zhao S, Ma C, Cui J, Zheng Y. Sulforaphane protects against cardiovascular disease via Nrf2 activation. Oxid Med Cell Longevity 2015; 2015: 407580.
Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol 2017; 12: 311–24.
Yan T, Yu X, Sun X, Meng D, Jia JM. A new steroidal saponin, furotrilliumoside from Trillium tschonoskii inhibits lipopolysaccharide-induced inflammation in Raw264.7 cells by targeting PI3K/Akt, MARK and Nrf2/HO-1 pathways. Fitoterapia 2016; 115: 37–45.
Shen WC, Liang CJ, Huang TM, Liu CW, Wang SH, Young GH, et al. Indoxyl sulfate enhances IL-1beta-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFkappaB/AP-1 pathway. Arch Toxicol 2016; 90: 2779–92.
Subhan F, Kang HY, Lim Y, Ikram M, Baek SY, Jin S, et al. Fish scale collagen peptides protect against CoCl2/TNF-alpha-induced cytotoxicity and inflammation via inhibition of ROS, MAPK, and NF-kappaB pathways in HaCaT cells. Oxid Med Cell Longevity 2017; 2017: 9703609.
Li C, Ma D, Chen M, Zhang L, Zhang L, Zhang J, et al. Ulinastatin attenuates LPS-induced human endothelial cells oxidative damage through suppressing JNK/c-Jun signaling pathway. Biochem Biophys Res Commun 2016; 474: 572–8.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 31671437), and the Natural Science Foundation of Guangdong Province, China (No 2016A030313335).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, St., Dai, Q., Zhang, Sx. et al. Ulinastatin attenuates LPS-induced inflammation in mouse macrophage RAW264.7 cells by inhibiting the JNK/NF-κB signaling pathway and activating the PI3K/Akt/Nrf2 pathway. Acta Pharmacol Sin 39, 1294–1304 (2018). https://doi.org/10.1038/aps.2017.143
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.143
Keywords
This article is cited by
-
Oral pyroptosis nanoinhibitor for the treatment of inflammatory bowel disease
Nano Research (2024)
-
Efficacy of urinary trypsin inhibitor in the treatment of rats with severe sepsis and its effects on coagulation and immunity
Molecular & Cellular Toxicology (2023)
-
Caudatin attenuates inflammatory reaction by suppressing JNK/AP-1/NF-κB/caspase-1 pathways in activated HMC-1 cells
Food Science and Biotechnology (2023)
-
Changes in Intracellular and Extracellular Metabolites of Mixed Lactobacillus Strains Enhance Inhibition of Pathogenic Bacterial Growth and Lipopolysaccharide-Induced Alleviation of RAW264.7 Cellular Inflammation
Probiotics and Antimicrobial Proteins (2023)
-
Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics
Inflammation (2023)
