
Abstract
Aim:
Fragment-based lead discovery (FBLD) is a complementary approach in drug research and development. In this study, we established an NMR-based FBLD platform that was used to screen novel scaffolds targeting human bromodomain of BRD4, and investigated the binding interactions between hit compounds and the target protein.
Methods:
1D NMR techniques were primarily used to generate the fragment library and to screen compounds. The inhibitory activity of hits on the first bromodomain of BRD4 [BRD4(I)] was examined using fluorescence anisotropy binding assay. 2D NMR and X-ray crystallography were applied to characterize the binding interactions between hit compounds and the target protein.
Results:
An NMR-based fragment library containing 539 compounds was established, which were clustered into 56 groups (8–10 compounds in each group). Eight hits with new scaffolds were found to inhibit BRD4(I). Four out of the 8 hits (compounds 1, 2, 8 and 9) had IC50 values of 100–260 μmol/L, demonstrating their potential for further BRD4-targeted hit-to-lead optimization. Analysis of the binding interactions revealed that compounds 1 and 2 shared a common quinazolin core structure and bound to BRD4(I) in a non-acetylated lysine mimetic mode.
Conclusion:
An NMR-based platform for FBLD was established and used in discovery of BRD4-targeted compounds. Four potential hit-to-lead optimization candidates have been found, two of them bound to BRD4(I) in a non-acetylated lysine mimetic mode, being selective BRD4(I) inhibitors.
Similar content being viewed by others
Introduction
Fragment-based lead discovery (FBLD) is becoming an indispensable alternative approach for drug development1,2. Although only a small number (500–5000) of fragment molecules are used to establish the FBLD library, the chemical space that they cover is comparable to that of conventionally used compound libraries containing tens of thousands of molecules. Moreover, because only a small number of molecules are incorporated, the efficiency of targeted screening is significantly enhanced. In practice, biophysics techniques, including NMR, MS and SPR etc, are applied to screen for hit compounds that bind to target proteins (drug targets). Next, the structure of the protein-hit complex is solved with either X-ray crystallography or NMR3,4,5. Using the solved structure of the complex, the hit compounds are further optimized to enhance binding and druggability. As mentioned above, NMR technology plays an important role in FBLD, both in screening the fragment library and in identifying the binding interactions. To improve the efficiency of drug discovery, here we report the successful establishment of an NMR platform for FBLD and demonstrate its application with the discovery of compounds targeting an important epigenetic drug target, BRD4.
Epigenetic traits are heritable variations in the patterns of post-translational modifications on histone proteins and in the methylation of DNA6,7,8,9,10,11,12,13. Changing an epigenetic modification pattern can switch genes on and off, thus affecting cellular functions, and dysfunction of epigenetic regulation contributes to the development of multiple human diseases14,15. Three classes of key epigenetic factors are writers, erasers and readers, which deposit, remove and recognize epigenetic modifications, respectively. Our target protein, BRD4, belongs to the BET subfamily of the bromodomain-containing protein family and contains two N-terminal bromodomains that read histone acetylation modifications16,17. The human genome encodes 61 bromodomains, which are present in 46 proteins and are classified into eight distinct subfamilies16,18. Dysfunction of bromodomains, including the members of the BET subfamily, have been reported to play important roles in the development of several aggressive types of cancer19. For example, the bromodomains of BRD4 promote human squamous carcinoma by forming a highly oncogenic fusion with NUT (nuclear protein in testis)20. Bromodomain proteins are promising epigenetic therapy targets for anti-cancer drug discovery and have already attracted extensive attention from medicinal chemists, in the growing field of bromodomain inhibitor discovery21. A large number of chemical scaffolds targeting bromodomains have been published16,22,23,24,25,26. However, none of them have been approved as cancer therapies, and additional effort should be applied in this field.
In this paper, we applied an NMR-based screening method to search for novel chemical scaffolds targeting the first bromodomain of BRD4 (hereafter referred to as BRD4(I)) and identified seven novel scaffolds. By characterizing complexes of BRD4(I) and the hit compounds, the interactions between BRD4(I) and hit compounds were revealed, and the structure-activity relationships for several of the hit fragments were elucidated. Our data provide new information for BRD4-targeted drug lead discovery.
Materials and methods
Fragment compound library
The fragment library is an essential component of fragment-based lead discovery, and its quality determines hit identification probability as well as lead druggability. Over the past decade, several strategies for constructing a fragment library have been proposed. The most familiar concept used in FBLD is the 'Rule of Three'. To generate our own fragment library, all of the small compounds in ZINC database were downloaded from the web site http://zinc.docking.org and filtered according to the following rules:
1. Molecular weight ≤ 300 Da
2. Rotatable bonds ≤ 5
3. logP ≤ 3.5
4. 1 ≤ smallest set of smallest ring ≤ 4.
Then, the resulting fragments were further clustered into groups with a Tanimoto similarity of 0.7 as the cutoff by using Pipeline Pilot software (version 7.5). Subsequently, the compounds labeled as the cluster center were selected as representatives of these clusters. To achieve a high diversity in the fragment library, only cluster-center compounds were selected and sent to two chemical vendors, Chemdiv and Enamine, for purchase inquiry. Finally, 800 fragments were purchased.
The 1H-NMR spectra of these 800 compounds were acquired for water solubility determination and group (compound mixture) generation. 4-Aminobenzoic acid (also known as para-aminobenzoic acid or PABA), which exhibits good water solubility, was chosen as the reference compound for water solubility assessment. To prepare the standard curve, the 1D 1H NMR spectra of PABA at concentrations of 50 μmol/L, 100 μmol/L, 200 μmol/L, 500 μmol/L and 1 mmol/L in the screening buffer were recorded, and the resulting integration values derived from the NMR signals of the PABA aromatic hydrogen were calculated and normalized to the sum of the NMR resonance intensity of DMSO. The standard curve was achieved by plotting the concentrations of PABA (horizontal axis) against the normalized integration values of its NMR signals (vertical axis). Then, the 1D 1H NMR spectra for each compound at a calculated concentration of 200 μmol/L in the screening buffer were acquired. The actual experimental concentrations of these fragment compounds, which were closely related to their water solubility, were determined by fitting the normalized integration data for the compounds to the established standard curve. Compounds with water solubility less than 100 μmol/L in PBS buffer (20 mmol/L NaH2PO4/Na2HPO4, 100 mmol/L NaCl, 2% DMSO, pH 7.4) were excluded from the library. To simplify library screening, the remaining 539 compounds were clustered into 56 groups (8–10 compounds in each group) by following the rule of no significant NMR signal overlap in the spectra of mixed group compounds. For all of the 56 groups, DMSO-d6 stock solutions with a group compound concentration of 10 mmol/L were prepared and used for screening.
Protein sample preparation
Protein samples of the first bromodomain of BRD4 (BRD4(I): BRD4 N-terminal fragment spanning N44 to E168) were prepared as previously described27,28. His-tagged BRD4(I) was expressed in Escherichia coli [BL21(DE3) competent cells] and purified by using a combination of affinity chromatography (Ni-NTA column) and size exclusion chromatography (HiLoad 16/600 Superdex 75 pg column) on an FPLC system. The FPLC fractions of BRD4(I) were concentrated and used in enzymatic assays, for crystallization and for NMR data collection. 15N- and 13C-labeled samples were produced by growth in M9 minimal medium with 15N-labeled ammonium chloride and 13C-labeled glucose as the nitrogen source and the carbon source, respectively.
NMR spectroscopy
All of the NMR data for BRD4(I) with or without the hit compounds were collected on a Bruker Avance III 600 MHz NMR spectrometer equipped with a cryogenically cooled probe at 25 °C.
Two-dimensional [1H, 15N] HSQC experiments were recorded on uniformly 15N-labeled BRD4(I) with or without the addition of a 10-fold molar excess of the hit compounds. A series of 3D triple-resonance spectra including the [1H, 15N, 13C] HNCA/HN(CO)CA pair, the [1H, 15N, 13C] HNCO/HN(CA)CO pair, and the [1H, 15N, 13C] HNCACB/CACB(CO)NH pair, which were acquired on 100% 15N and 100% 13C double-labeled BRD4(I) in its free state, were used to obtain the backbone chemical shift assignments of the protein. The concentrations of BRD4(I) for the 2D [1H, 15N] HSQC spectra and the 3D triple-resonance experiments were 0.05 and 0.6 mmol/L, respectively.
NMR data analysis
NMR data processing and analysis were performed using the programs NMRPipe29, CARA30, and Sparky (Goddard and Kneller, Sparky 3, University of California, San Francisco). The chemical shift perturbation values (Δδavg) for 15N and 1H nuclei were derived from equation (1):

in which ΔδN and ΔδH represent the chemical shift perturbation value of the amide nitrogen and proton, respectively.
Hit compound screening
Two cycles of BRD4(I)-targeted hit compound screening were performed using ligand-based T1ρ and saturation transfer difference (STD) NMR experiments. In the first round of screening, grouped fragment compound samples containing 200 μmol/L of the compound mixture (8–10 fragment compounds in each group) or 200 μmol/L compound mixture in the presence of 20 μmol/L protein, were dissolved in phosphate buffer and used for NMR data acquisition. The identified potential hit compound candidates were then subjected to a second round of screening with STD and T1ρ NMR experiments. The samples used in the second round of screening were 200 μmol/L potential hit compound or 200 μmol/L potential hit compound in the presence of 5 μmol/L protein. All of the ligand-based T1ρ and STD NMR experiments were performed at 25 °C on a Bruker Avance III 600 MHz NMR spectrometer equipped with a cryogenically cooled probe.
Crystallization and data collection
Aliquots of purified BRD4(I) protein were prepared for crystallization using the vapor diffusion method. Crystals were grown by mixing 1 μL of the protein (9 mg/mL) with an equal volume of reservoir solution containing 6 mol/L sodium formate and 10% glycerol (Compound 1); 25% PEG 6000, 0.1 mol/L Tris, 0.2 mol/L MgCl2, pH 8.0 (Compound 6); or 20% PEG 3350, 0.2 mol/L NaNO3, 0.1 mol/L bis-Tris-propane, pH 8.5 (Compound 9). Crystals grew to diffracting quality within 1–3 weeks in all of the cases.
Data were collected at 100 K on the beamline BL17U at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). The data were processed with XDS software packages, and the structures were solved using PHASER2.3.0. The search model used for molecular replacement was 4QR3 from the Protein Data Bank (PDB). All of the structures were refined using PHENIX. With the aid of the program Coot, the compound and water were fitted into the initial Fo–Fc maps. The complete statistics, as well as the quality of the solved structures, are shown in Supporting Information Table S1. The structures have been deposited in the PDB under the deposition codes 5HQ5, 5HQ6 and 5HQ7. The analysis of BRD4(I)-hit compound co-crystal structures were performed with LigPlot+31.
Fluorescence anisotropy binding assay
The binding affinities of the hit compounds for BRD4(I) were assessed by using a fluorescence anisotropy binding assay as described previously27,28. All of the components were dissolved in a buffer containing 50 mmol/L HEPES, 150 mmol/L NaCl and 0.5 mmol/L CHAPS at pH 7.4 with final concentrations of 20 nmol/L BRD4(I) and 5 nmol/L fluorescent ligand. The test compound in step-wise concentration series or the DMSO vehicle and the above-mentioned reaction mixture were added into a Corning 384-well black low volume plate (CLS3575) and equilibrated in the dark for 17 h at 4 °C. Fluorescence anisotropy was read on a BioTek Synergy2 multimode microplate reader (λex=485 nm, λem=530 nm; dichroic, 505 nm).
Results and discussion
Fragment library
To increase the efficiency of drug discovery, a combination of random screening and structure-based rational drug design is applied during FBLD. Biophysical techniques, including NMR, X-ray crystallography, and SPR, are the most commonly used screening and/or protein-compound complex characterization approaches in FBLD32. In this paper, we sought to establish an NMR-based fragment library for FBLD. Initially, 800 commercially available fragment compounds were selected by following the “Rule of Three” and were purchased. Next, the solubility of each compound in screening buffer (20 mmol/L NaH2PO4/Na2HPO4, 100 mmol/L NaCl, 2% DMSO-d6, pH 7.4) was determined with 1D 1H NMR spectroscopy. A total of 261 compounds with water solubility values less than 100 μmol/L were excluded from the library. To simplify library screening, the remaining 539 compounds were clustered into 56 groups (8–10 compounds in each group) by following the rule of no significant NMR signal overlap in the spectra of mixed group compounds (Supporting Information Figure S1).
Hit generation
Ligand-based NMR approaches (T1ρ, saturation transfer difference–STD, and WaterLOGSY, among others) and target-based NMR methods ([1H, 15N] HSQC and [1H, 13C] HSQC) are two major classes of NMR techniques that are commonly used for the primary NMR screening of hit compounds5. In comparison with ligand-based 1D NMR approaches, which can only assess whether the ligand actually binds to the target, the target-based NMR methods are more time-consuming but can provide additional information to identify where the ligand binds on the target33,34. Here, the ligand-based T1ρ and saturation transfer difference NMR experiments were applied to screen for BRD4-targeted hit compounds. After the primary group screening and the second round of single molecule evaluation, ten hits including (6,7-dimethoxy-quinazolin-4-yl)-ethyl-amine (compound 1), 2-chloro-6,7-dimethoxy-quinazolin-4-ylamine (compound 2), (3,5-difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (compound 3), (1-furan-2-yl-ethyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (compound 4), 4-methyl-1,3-dihydro-benzo[b][1,4]diazepin-2-one(compound 5), 2,6-dimethyl-4H-benzo[1,4]oxazin-3-one(compound 6), 2',4'-dihydro-1'H-spiro[cyclohexane-1,3'-isoquinolin]-1'-one (compound 7), cyclopropyl-(3-ethyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-amine (compound 8), (7-methyl-[1,2,4]triazolo[4,3-a]pyrimidin-5-yl)-o-tolyl-amine (compound 9) and 4-benzylsulfanyl-5H-pyrrolo[3,2-d]pyrimidine (compound 10) were identified (Figure 1, Supporting Information Figure S2–S11). All of these ten hit compounds, except compound 3 and 4, which share the same [1,2,4]triazolo[1,5-a]pyrimidin core structure reported to interact with BRD4(I)35, were identified to have novel scaffolds that down-regulated BRD4.

Chemical structure of ten fragments identified as BRD4(I) hit compounds. Of these ten hit compounds, compound 1 and compound 2 share a quinazolin scaffold, and compound 3 and compound 4 share a [1,2,4]triazolo[1,5-a]pyrimidin core structure.
Inhibition activity of Hit compounds
Two cycles of the fluorescence anisotropy binding assay were performed to determine the inhibition activities of the ten hit compounds on BRD4(I). The first cycle of the binding assay tested the inhibition rates of the 10 compounds at a concentration of 100 μmol/L. The compounds (compounds 1, 2, 6, 8, and 9) clearly inhibiting BRD4(I) were identified. In the second round of experiments, the IC50 values for compounds 1, 6, and 8 were determined to be in the hundreds of micromolar range (Figure 2). However, due to the low solubility of compound 2, its IC50 value was not determined, although this compound did show an inhibitory activity comparable to that of compound 1 in the first-round fluorescence anisotropy binding assay. For compound 9, which exhibited a similar inhibitory activity to compounds 1 and 2, the IC50 value was not determined due to its unexpectedly low stability. The inhibition activity data for four of the hit compounds (compounds 1, 2, 8, and 9) suggest that they are promising candidates for further BRD4-targeted hit-to-lead optimization.

Inhibitory activities of compound 1, compound 6 and compound 8 on BRD4(I) determined with fluorescence anisotropy binding assays. Quantification plots of the fluorescence anisotropy binding assays for hit compounds 1, 6 and 8 are presented in A, B and C, respectively.
Backbone resonance assignments of BRD4(I)
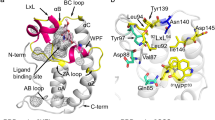
The classical strategy was applied to obtain the backbone resonance assignments of BRD4(I). In total, 105 non-proline residues were identified (Figure 3). Based on the backbone resonance assignment data, the secondary structural elements of BRD4(I) in solution were predicted according to the chemical shift index analysis (CSI) results36,37. The consensus CSI (Cα) output suggested a canonical bromodomain global fold containing four α-helices (helix Z: Q64–W75, helix A: M107–E115, helix B: A122-C136, helix C: I146–Q159) (Figure 3)38. The NMR data indicated that the global solution structure of BRD4(I) showed no significant differences from its crystal structure.

NMR-based characterization of the solution structure of BRD4(I). (A) [1H, 15N] HSQC spectrum of BRD4(I). Backbone amide resonance assignments are labeled with the one-letter amino acid code and the sequence number. The insert shows an expanded view of a region with cross-peaks partially overlapped. (B) Consensus chemical shift index (Cα) for BRD4(I). The predicted secondary structural elements are shown together with the CSI plot. (C) Ribbon representation of the crystal structure of BRD4(I) (PDB code: 2OSS).
Characterization of BRD4(I)-Hit compound interactions
It has been well established that bromodomains share a conserved global fold composed of a left-handed bundle of four α-helices (αZ, αA, αB and αC, Figure 3) that are linked by diverse ZA and BC loop regions (Figure 3)38. The aromatic and hydrophobic residues in the ZA and BC loops form a recognition pocket for endogenous bromodomain ligands, acetyl-lysine histones. When acetylated lysine on an acetyl-lysine histone binds to the bromodomain, it becomes anchored to a conserved asparagine residue (N140 in BRD4(I)) through a hydrogen bond between the acetyl moiety and the sidechain of the asparagine residue, and extensive hydrophobic interactions between the acetylated lysine and the hydrophobic cavity of bromodomains occur16,38. To date, all of the bromodomain inhibitors developed competitively bind to the endogenous ligand recognition pocket. Based on the presence or absence of moieties that act as acetylated lysine mimetics, these inhibitors can be classified into two groups. The bromodomain inhibitors in the non-acetylated lysine mimetic group interact but do not form the canonical hydrogen bond with the conserved asparagines (N140 in BRD4(I)), whereas the other class of bromodomain inhibitors directly engages the protein module by forming the canonical hydrogen bond16. To categorize the novel BRD4-targeted scaffolds and extract the structure-activity relationship information, we characterized the BRD4(I)-hit compound interactions with biophysical techniques, including NMR and X-ray crystallography.
First, the NMR [1H,15N]HSQC experiments were used to characterize the interactions between hit compounds and BRD4(I). The [1H, 15N]HSQC spectrum commonly serves as a “fingerprint” of the protein backbone. Each 1H-15N cross peak in the HSQC spectrum represents a resonance peak from a single HN group on a specific amino acid residue in the protein (Figure 3). Because the chemical shift value at which an atom resonates is sensitive to its chemical environment39,40,41, amino acid residue-specific CSP are observed in the [1H, 15N] HSQC spectrum of a target protein upon ligand binding. Protein residues at contact surfaces or structural changes to the target protein induced by ligand binding can be identified through CSP analysis of the target protein upon titration of its binding partners (ligands). For BRD4(I)-hit compound (compounds 1, 6, 7, 8, 9, and 10) complexes, the CSP analysis was performed by two dimensional [1H, 15N] HSQC on uniformly 15N-labeled BRD4(I) with or without the addition of a 10-fold molar excess of the hit compounds (Figure 4, Supporting Information Figure S12–S17). The BRD4(I) residues significantly perturbed upon the binding of hit compounds were then identified (Figure 4). These residues clearly grouped into the ZA-loop segment (K76 to D106) and the BC-loop region (Y137 to A150) of BRD4(I) (Figure 3, Figure 4), indicating that all of these hit compounds bind to the endogenous ligand recognition pocket of the target protein. Moreover, the extremely hydrophobic residue W81 in BRD4, which is in the outer region of the ligand binding cavity, showed significant CSP with the addition of compounds 8, 9 or 10. This suggests that hit compounds 8, 9 and 10 might occupy a wider space when interacting with BRD4(I), and this observation might be attributed to the bulky moieties attached to the core structures of these three compounds (Figure 1).

Chemical shift perturbation (CSP) analysis for BRD4(I) upon hit compound binding. Solid and dashed lines indicate the mean and mean±SD values, respectively. Residues with CSP values above the dashed line are labeled. (A) CSP analysis for BRD4(I) after binding of hit compound 1. (B) CSP analysis for BRD4(I) after binding of hit compound 6. (C) CSP analysis for BRD4(I) after binding of hit compound 7. (D) CSP analysis for BRD4(I) after binding of hit compound 8. (E) CSP analysis for BRD4(I) after binding of hit compound 9. (F) CSP analysis for BRD4(I) after binding of hit compound 10.
We sought to characterize the interactions between hit compounds and the target protein by solving the structures of the complexes. After extensive effort, the crystal structures for the complexes of BRD4(I)-compound 1, BRD4(I)-compound 6 and BRD4(I)-compound 9 were successfully determined (Figure 5, Supporting Information Table S1). According to the structures, two of the three hits (compound 6 and compound 9) are acetylated lysine mimetic inhibitors, and compound 1 is a non-acetylated lysine mimetic inhibitor (Figure 5). Interestingly, although it has been reported that non-acetylated lysine mimetic inhibitors usually exhibit weak binding affinities for bromodomains16,42,43,44,45, compound 1 showed moderate inhibitory activity toward BRD4(I) (Figure 2). This observation might be explained by the interaction plot data extracted from the complex structure of BRD4(I)-compound 1 (Figure 5), which suggest that compound 1 fits well into the ligand binding pocket of BRD4(I), and extensive hydrophobic interactions are formed between the compound and the target protein (Figure 5). The scaffold related to compounds 1 and 2 might represent a good starting structure for development of non-acetylated lysine mimetic inhibitors. Of the two acetylated lysine mimetic inhibitors (compound 6 and compound 9), compound 9 showed stronger binding affinity and more hydrophobic interactions (Figure 5). We believe that these results might be attributed to the additional hydrophobic interactions formed between the methyl benzyl group of compound 9 attached to its core structure and the WPF shelf (W81, P82 and F83) of BRD4(I) (Figure 4, Figure 5). However, because compound 8, which has a smaller chemical moiety attachment, exhibited the largest inhibitory activity towards BRD4(I) (Figure 2), changing the ring size of the methyl benzyl group in compound 9 might enhance its inhibitory activity.

Expanded view of BRD4(I)-hit compound co-crystal structures and schematic diagrams of BRD4(I)-hit compound interactions. (A) Expanded view of BRD4(I) bound to hit compound 1. (B) Schematic diagram of BRD4(I)-hit compound 1 interactions. Extensive hydrophobic interactions form between BRD4(I) and hit compound 1, and the involved residues in BRD4(I) and the atoms in hit compound 1 are highlighted with spiked lines. (C) Expanded view of BRD4(I) bound to hit compound 6. (D) Schematic diagram of BRD4(I)-hit compound 6 interactions. The residues in BRD4(I) and the atoms in hit compound 6, which are involved in the hydrophobic interaction network of these two molecules, are highlighted with spiked lines. The hydrogen bonds are highlighted with green dashed lines, and the blue spheres represent water molecules. (E) Expanded view of BRD4(I) bound to hit compound 9. (F) Schematic diagram of BRD4(I)-hit compound 9 interactions. The residues in BRD4(I) and the atoms in hit compound 9, which are involved in the hydrophobic interaction network of these two molecules, are highlighted with spiked lines. The hydrogen bonds are highlighted with green dashed lines, and the blue spheres represent water molecules.
In summary, FBLD and design has become a promising complementary approach for drug development. In this paper, we report the successful establishment of an NMR-based fragment library and research platform for FBLD studies in our institute. Utilizing our NMR-based FBLD platform, seven BRD4-targeted novel scaffolds and the related structure-activity relationship information were successfully obtained. Moreover, the inhibitory activities of compound 1 and compound 8 on BRD4(I) had IC50 values of 265.54 μmol/L and 106.62 μmol/L, respectively. Compounds 2 and 9 showed a similar inhibitory activity to that of compound 1 in the first round of fluorescence anisotropy binding assays (their IC50 values could not be accurately determined due to either the low solubility or the low stability of the compounds). The inhibition activity data indicate that compounds 1, 2, 8 and 9 have potential for further BRD4-targeted hit-to-lead optimization. It is worth of noting that of the four candidates, compounds 1 and 2 share a common quinazolin core structure and bind to BRD4(I) in a non-acetylated lysine mimetic mode. Because non-acetylated lysine mimetic inhibitors have the capability to be selective BRD4 inhibitors, our results provide a basis for the development of different types of BRD4 inhibitors.
Author contribution
Nai-xia ZHANG, Bing XIONG, Ye-chun XU, and Jing-kang SHEN designed the experiments. Nai-xia ZHANG, Bing XIONG, Tian-tian CHEN and Jun-lan YU wrote the main manuscript text. Jun-lan YU, Chen ZHOU, Xu-long TANG, Fu-lin LIAN, and Yi WEN performed the NMR experiments and the related data analysis. Jun-lan YU and Tian-tian CHEN solved the crystal structures of the protein-hit compound complexes. Jun-lan YU performed the fluorescence anisotropy binding assay. All of the authors reviewed the manuscript.
Accession codes
References
Erlanson DA . Introduction to fragment-based drug discovery. Top Curr Chem 2012; 317: 1–32.
Congreve M, Chessari G, Tisi D, Woodhead AJ . Recent developments in fragment-based drug discovery. J Med Chem 2008; 51: 3661–80.
Hanzawa H, Takizawa T . NMR in the fragment-based drug discovery. Tanpakushitsu Kakusan Koso 2009; 54 (12 Suppl): 1617–21. Japanese.
Harner MJ, Frank AO, Fesik SW . Fragment-based drug discovery using NMR spectroscopy. J Biomol NMR 2013; 56: 65–75.
Campos-Olivas R . NMR screening and hit validation in fragment based drug discovery. Curr Top Med Chem 2011; 11: 43–67.
Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B . Targeting BET bromodomains for cancer treatment. Epigenomics 2015; 7: 487–501.
la Rosa AH, Acker M, Swain S, Manoharan M . The role of epigenetics in kidney malignancies. Cent Eur J Urol 2015; 68: 157–64.
Laffita-Mesa JM, Bauer PO, Kouri V, Pena Serrano L, Roskams J, Almaguer Gotay D, et al. Epigenetics DNA methylation in the core ataxin-2 gene promoter: novel physiological and pathological implications. Hum Genet 2012; 131: 625–38.
Laird PW, Jaenisch R . The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet 1996; 30: 441–64.
Ledford H . Epigenetics: The genome unwrapped. Nature 2015; 528: S12–3.
Lu Q, Qiu X, Hu N, Wen H, Su Y, Richardson BC . Epigenetics, disease, and therapeutic interventions. Ageing Res Rev 2006; 5: 449–67.
Lund AH, van Lohuizen M . Epigenetics and cancer. Genes Dev 2004; 18: 2315–35.
Lundstrom K . What is the potential of epigenetics in drug development? Future Med Chem 2015; 7: 239–42.
Zhao H, Ning S, Scicinski J, Oronsky B, Knox SJ, Peehl DM . Epigenetic effects of RRx-001: a possible unifying mechanism of anticancer activity. Oncotarget 2015; 6: 43172–81.
Huang CZ, Yu T, Chen QK . DNA methylation dynamics during differentiation, proliferation, and tumorigenesis in the intestinal tract. Stem Cells Dev 2015; 24: 2733–9.
Filippakopoulos P, Knapp S . Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 2014; 13: 337–56.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010; 468: 1067–73.
Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012; 149: 214–31.
Belkina AC, Denis GV . BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012; 12: 465–77.
French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA . BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res 2003; 63: 304–7.
Smith SG, Zhou MM . The bromodomain: a new target in emerging epigenetic medicine. ACS Chem Biol 2016; 11: 598–608.
Xue X, Zhang Y, Liu Z, Song M, Xing Y, Xiang Q, et al. Discovery of benzo[cd]indol-2(1H)-ones as potent and specific BET bromodomain inhibitors: structure-based virtual screening, optimization, and biological evaluation. J Med Chem 2016; 59: 1565–79.
Hugle M, Lucas X, Weitzel G, Ostrovskyi D, Breit B, Gerhardt S, et al. 4-Acyl pyrrole derivatives yield novel vectors for designing inhibitors of the acetyl-lysine recognition site of BRD4(1). J Med Chem 2016; 59: 1518–30.
Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, et al. Exploring selective inhibition of the first bromodomain of the human bromodomain and extra-terminal domain (BET) proteins. J Med Chem 2016; 59: 1634–41.
Ran X, Zhao YJ, Liu L, Bai LC, Yang CY, Zhou B, et al. Structure-based design of gamma-carboline analogues as potent and specific BET bromodomain inhibitors. J Med Chem 2015; 58: 4927–39.
McKeown MR, Shaw DL, Fu H, Liu S, Xu X, Marineau JJ, et al. Biased multicomponent reactions to develop novel bromodomain inhibitors. J Med Chem 2014; 57: 9019–27.
Zhao L, Cao D, Chen T, Wang Y, Miao Z, Xu Y, et al. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J Med Chem 2013; 56: 3833–51.
Zhao L, Wang Y, Cao D, Chen T, Wang Q, Li Y, et al. Fragment-based drug discovery of 2-thiazolidinones as BRD4 inhibitors: 2. Structure-based optimization. J Med Chem 2015; 58: 1281–97.
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A . NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995; 6: 277–93.
Keller R . The computer aided resonance assignment tutorial. Germany: CANTINA Verlag; 2004.
Laskowski RA, Swindells MB . LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011; 51: 2778–86.
Hubbard RE, Murray JB . Experiences in fragment-based lead discovery. Methods Enzymol 2011; 493: 509–31.
Shi L, Wen Y, Zhang N . 1H, 13C and 15N backbone and side-chain resonance assignments of the N-terminal ubiquitin-binding domains of USP25. Biomol NMR Assign 2014; 8: 255–8.
Gao J, Ma R, Wang W, Wang N, Sasaki R, Snyderman D, et al. Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex. PLoS One 2014; 9: e 88098.
Vidler LR, Filippakopoulos P, Fedorov O, Picaud S, Martin S, Tomsett M, et al. Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening. J Med Chem 2013; 56: 8073–88.
Andersen NH, Cao B, Chen C . Peptide/protein structure analysis using the chemical shift index method: upfield alpha-CH values reveal dynamic helices and alpha L sites. Biochem Biophys Res Commun 1992; 184: 1008–14.
Wishart DS, Sykes BD, Richards FM . The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992; 31: 1647–51.
Filippakopoulos P, Knapp S . The bromodomain interaction module. FEBS Lett 2012; 586: 2692–704.
Williamson MP . Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc 2013; 73: 1–16.
Wen Y, Shi L, Ding Y, Cui R, He WT, Hu HY, et al. The N-terminal ubiquitin-binding region of ubiquitin-specific protease 28 modulates its deubiquitination function: NMR structural and mechanistic insights. Biochem J 2015; 471: 155–65.
Zhang H, Zhou C, Chen W, Xu Y, Shi Y, Wen Y, et al. A dynamic view of ATP-coupled functioning cycle of Hsp90 N-terminal domain. Sci Rep 2015; 5: 9542.
Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem Biol 2011; 18: 531–41.
Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, et al. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem Biol 2006; 13: 81–90.
Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, et al. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem 2013; 56: 9251–64.
Ito T, Umehara T, Sasaki K, Nakamura Y, Nishino N, Terada T, et al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem Biol 2011; 18: 495–507.
Acknowledgements
NMR data were acquired in the NMR facility of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences. X-ray crystallography data were recorded in the Shanghai Synchrotron Radiation Facility. This work was funded by grants from the National Natural Science Foundation of China (21272246 to Nai-xia ZHANG, 31300608 to Yi WEN, and 81330076 to Jing-kang SHEN) and the National Key Basic Research Program of China (2013CB910900 to Nai-xia ZHANG).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available on the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Figure S1
Illustration of the principles for group generation of fragment compounds. (DOC 1184 kb)
Supplementary Figure S2
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 1 directly interacts with BRD4(I). (DOC 113 kb)
Supplementary Figure S3
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 2 directly interacts with BRD4(I). (DOC 101 kb)
Supplementary Figure S4
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 3 directly interacts with BRD4(I). (DOC 93 kb)
Supplementary Figure S5
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 4 directly interacts with BRD4(I). (DOC 85 kb)
Supplementary Figure S6
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 5 directly interacts with BRD4(I). (DOC 97 kb)
Supplementary Figure S7
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 6 directly interacts with BRD4(I). (DOC 99 kb)
Supplementary Figure S8
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 7 directly interacts with BRD4(I). (DOC 100 kb)
Supplementary Figure S9
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 8 directly interacts with BRD4(I). (DOC 88 kb)
Supplementary Figure S10
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 9 directly interacts with BRD4(I). (DOC 1872 kb)
Supplementary Figure S11
Ligand observed T1ρ and saturation transfer difference (STD) spectra indicate that compound 10 directly interacts with BRD4(I). (DOC 99 kb)
Supplementary Figure S12
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 1 (black, molar ratio of 1:10 BRD4(I) to compound 1) reveals spectral changes upon hit compound binding. (DOC 85 kb)
Supplementary Figure S13
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 6 (black, molar ratio of 1:10 BRD4(I) to compound 6) reveals spectral changes upon hit compound binding. (DOC 83 kb)
Supplementary Figure S14
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 7 (black, molar ratio of 1:10 BRD4(I) to compound 7) reveals spectral changes upon hit compound binding. (DOC 79 kb)
Supplementary Figure S15
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 8 (black, molar ratio of 1:10 BRD4(I) to compound 8) reveals spectral changes upon hit compound binding. (DOC 92 kb)
Supplementary Figure S16
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 9 (black, molar ratio of 1:10 BRD4(I) to compound 9) reveals spectral changes upon hit compound binding. (DOC 85 kb)
Supplementary Figure S17
Superposition of [1H, 15N] HSQC spectra of BRD4(I) without (red) and with compound 10 (black, molar ratio of 1:10 BRD4(I) to compound 10) reveals spectral changes upon hit compound binding. (DOC 84 kb)
Supplementary Table S1
Structural statistics of BRD4(I)-hit compound co-crystal structures. (DOC 48 kb)
Rights and permissions
About this article

Cite this article
Yu, Jl., Chen, Tt., Zhou, C. et al. NMR-based platform for fragment-based lead discovery used in screening BRD4-targeted compounds. Acta Pharmacol Sin 37, 984–993 (2016). https://doi.org/10.1038/aps.2016.19
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.19
