
Abstract
The signal transducer and activator of transcription 3 (STAT3) plays a critical role in platelet functions. This study sought to understand the effects of the STAT3 inhibitor SC99 on platelet activation and aggregation. Immunoblotting assays were applied to measure the effects of SC99 on the STAT3 signaling pathway. A ChronoLog aggregometer was used to evaluate platelet aggregation. A flow cytometer was used to evaluate P-selectin expression in the presence of SC99. AlamarBlue and Annexin-V staining were used to evaluate platelet viability and apoptosis, respectively. A fluorescence microscope was applied to analyze platelet spreading. SC99 inhibited the phosphorylation of JAK2 and STAT3 in human platelets but had no effects on the phosphorylation of AKT, p65 or Src, all of which are involved in platelet activation. Further studies revealed that SC99 inhibited human platelet aggregation induced by collagen and thrombin in a dose-dependent manner. SC99 inhibited thrombin-induced P-selectin expression and fibrinogen binding to single platelets. Moreover, SC99 inhibited platelet spreading on fibrinogen and clot retraction mediated by outside-in signaling. SC99 inhibited platelet aggregation in mice but it did not significantly prolong the bleeding time. Taken together, the present study revealed that SC99 inhibited platelet activation and aggregation as a STAT3 inhibitor. This agent can be developed as a promising treatment for thrombotic disorders.
Similar content being viewed by others

Introduction
Thrombosis is a critical physiological process in the event of bleeding or injuries that prevents excessive blood loss by forming a blood clot in the vessels. Platelets are the central component in thrombus formation, in which platelet activation and aggregation are essential steps. In the platelet activation, several key molecular events have been clearly understood, including the phosphorylation of the Fc receptor γ-chain (FcRγ-chain)1; the formation of the glycoprotein GPIb-IX-V complex on platelet membranes2; and the activation of signaling proteins, such as Src, Syk, PLCγ2, phosphatidylinositol 3-kinase (PI3K) and AKT3. All of these signal cascades converge on platelet aggregation mediated by fibrinogen binding to the integrin αIIbβ3 and thrombus formation4,5,6,7. Therefore, targeting platelets is a pivotal strategy to prevent and treat thrombosis and embolic diseases, such as stroke and myocardial infarction. Several classes of drugs have been developed, including (1) aspirin and triflusal, which irreversibly inhibits the enzyme COX, resulting in reduced platelet production of TXA2; (2) clopidogrel, which affects the ADP-dependent activation of the IIb/IIIa complex; and (3) glycoprotein IIb/IIIa receptor antagonists, which block a receptor on the platelet for fibrinogen and von Willebrand factor8. These drugs generate considerable improvements in the management of cardiovascular diseases; however, the risk of uncontrolled bleeding of the known anti-platelet drugs limits their extensive clinical applications8. Vascular thrombosis results in 2.5 million deaths in China annually. In the USA, one American dies from stroke every 4 minutes9. Therefore, it is urgent to develop novel safe and effective anti-platelet drugs.
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor activated by cytokine-induced intracellular signals10,11. As a nuclear transcription factor, STAT3 plays an important role in megakaryopoiesis, platelet production and maturation. Specifically, thrombopoietin binds its receptor on megakaryocytes to activate the receptor-associated Janus kinase (JAK) and to recruit and activate STAT312,13. STAT3 subsequently changes its conformation, dimerizes through its SH2 domain, and translocates into the nuclei, where it regulates the transcription of multiple genes required for platelet production. Recent studies revealed that STAT3 also acts in a manner other than genomic regulation to modulate platelet activation and aggregation by interplaying with Syk and PLCγ214. Therefore, targeting JAK2/STAT3 signaling could be an emerging strategy in the management of platelet-associated diseases. Several inhibitors of the JAK2/STAT3 signaling pathway modulate platelet activation, including AG490 and TG10134815.
We recently identified a novel STAT3 inhibitor SC99 that displays potent anti-myeloma activity but exhibits no overt toxicity16. In the present study, we found that SC99 displays promising effects against platelet activation and aggregation. Given its undetectable toxicity, SC99 could be developed as an anti-platelet drug for the treatment of thrombosis-associated diseases, such as stroke and myocardial infarction.
Materials and methods
Reagents
SC99 was synthesized and purified by high-performance liquid chromatography (HPLC) as reported previously16. Collagen (Cat #385) and thrombin (Cat #386) were purchased from ChronoLog Corp (Havertown, PA, USA). Antibodies against STAT3 (Cat #9139L), phospho-STAT3 (Cat #9131L), phospho-Akt Thr308 (Cat #9275L), and phospho-Src (Cat #2101) were purchased from Cell Signaling Technologies (Danvers, MA, USA), phospho-p65 (Cat #2220-1) was purchased from Epitomics (Beijing, China), phycoerythrin (PE)-conjugated anti-human CD62P (Cat #550561) was purchased from BD Biosciences (San Jose, CA, USA). JAK2 (Cat #YM0385), and phospho-JAK2 (Cat #YP0155) antibodies were obtained from Immunoway (Suzhou, China). FITC-conjugated polyclonal anti-human fibrinogen (Cat #GF011129) was purchased from GeneTech (Shanghai, China), and normal mouse IgG/PE and normal rabbit IgG/FITC (Cat #sc-2866, sc-3870) were purchased from Santa Cruz Biotechnology. An Annexin V-FITC kit (Cat #BU-AP0103) was purchased from Biouniquer (Nanjing, China). Phalloidin-TRITC (Cat #p1951) was purchased from Sigma. HRP goat anti-mouse IgG (H+L) (Cat #A0216) and HRP goat anti-rabbit IgG (H+L) (Cat #A0208) were purchased from Beyotime, Nantong, China.
Platelet isolation
Human venous blood was obtained from healthy donors who provided informed consent. To obtain platelet-rich plasma, whole blood was treated with ACD buffer containing 65 mmol/L Na3 citrate, 70 mmol/L citric acid, 100 mmol/L dextrose, pH 4.4, followed by centrifuging at 900 rounds per minute for 20 min. After washing, the enriched platelets were resuspended, counted, and adjusted to 3×108/mL using HEPES-buffered Tyrode's solution containing 119 mmol/L NaCl, 5 mmol/L KCl, 25 mmol/L HEPES buffer, 2 mmol/L CaCl2, 2 mmol/L MgCl2, and 6 g/L glucose, pH 7.4.
Platelet aggregation
Platelets aggregation was performed in a ChronoLog aggregometer (Havertown, PA, USA). Briefly, platelets were pre-incubated with vehicle or SC99 at 37 °C in a cuvette. The aggregation assay was started with 0% aggregation baseline. Then, an agonist was added to observe the percentage of platelet aggregation with stirring at 900 rounds per minute in the presence of CaCl2 (1 mmol/L). To analyze the effects of SC09 on platelet aggregation in vivo, mice were administered ip SC99 (2 mg/kg) or vehicle. Forty-five min later, blood was obtained from the inferior vena cava, Platelets were isolated and stimulated with collagen (2 μg/mL) for 1 min followed by the evaluation of platelet aggregation. Three independent assays were performed.
Soluble fibrinogen binding and P-selectin expression
Platelets were pre-incubated with or without SC99 (5 μmol/L) for 10 min at 37 °C, followed by stimulation with or without thrombin (0.05 U/mL) in the presence of FITC-labeled fibrinogen or PE-labeled CD62P at 37 °C for 10 min. Platelet-bound fluorescence was analyzed using a flow cytometer (BD FACS Calibur™).
Platelet viability assay
A platelet viability assay with the AlamarBlue cell viability reagent was used as described previously17 and according to the manufacturer's instruction (Invitrogen, Carlsbad, CA, USA). Briefly, platelets (100 μL, 3×108/mL) were pre-incubated for 15 min with SC99 or vehicle followed by the addition of AlamarBlue reagent (10 μL) and incubation for an additional 4 h at 37 °C. Live platelets were then determined using a SpectraMax Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) with excitation and emission wavelengths of 570 and 585 nm, respectively.
Platelet apoptosis determination
Platelet apoptosis was determined via the external exposure of phosphatidylserine on the platelet membrane using Annexin-V and PI staining. Briefly, platelets (100 μL, 2×107/mL) were pre-incubated with SC99 or vehicle at 37 °C for 15 min followed by staining with Annexin V-FITC and PI and analysis on a flow cytometer (FACSCalibur™, Becton Dickinson, San Jose, CA, USA).
Immunoblotting
Aliquots of platelets (250 μL, 3×108/mL) were pre-incubated with SC99 or vehicle for 10 min and stimulated by agonists under stirring for 5 min at 37 °C. The reaction was stopped by adding RIPA buffer (1% Triton X-100, 1% deoxycholate, 0.1% SDS, 10 mmol/L Tris, and 150 mmol/L NaCl) containing protease inhibitors and phosphatase inhibitors. After being heated to 97 °C for 10 min, proteins were fractionated via 10% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Millipore). The membrane was incubated with specific antibodies at 4 °C overnight, followed by a secondary antibody (goat anti-rabbit IRDye 800CW or goat anti-mouse IRDye 800CW). The Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA) was used to analyze the signals.
Platelet spreading on immobilized fibrinogen
Glass coverslips were first coated with fibrinogen by soaking in 10 μg/mL fibrinogen in 0.1 mol/L NaHCO3 (pH 8.3) overnight at 4 °C. Simultaneously, platelets were pre-incubated with or without SC99 at 37 °C for 10 min. These platelets (2×106/mL in Tyrode's buffer) were added to the fibrinogen-immobilized coverslips and incubated for 60 min at room temperature. Free platelets were then washed away, and adherent platelets were fixed with 4% paraformaldehyde, followed by staining with TRITC-labeled phalloidin containing 0.1% Triton X-100 at room temperature for 2 h. The coverslips were mounted on the glass slides, and platelet spreading was analyzed under a fluorescence microscope (Olympus, FSX100).
Clot retraction
Platelet-rich plasma was incubated with SC99 (5 μmol/L) for 10 min at 37 °C. After that, platelet-rich plasma was dispensed in 0.25-mL aliquots into 5-mL tubes. Clot retraction was initiated by 0.08 U/mL of thrombin and allowed to proceed at 37 °C followed by monitoring at indicated time points using a digital camera. The extent of the retraction was quantified using Image J software developed by National Health Institute (Bethesda, MD, USA).
Tail bleeding time in mice
SC99 (2 mg/kg body weight) or vehicle was administered intraperitoneally to male mice (C57BL/6, 6–8 weeks old). Forty-five min later, mice were anesthetized with pentobarbital (100 mg/kg, ip) and placed on a heating pad from which the tail protruded. After the distal 5 mm of the tail was transected, the tail was immediately immersed in 12 mL of 0.9% sodium chloride for 10 min at 37 °C, and the time to bleeding cessation was recorded.
Animals and human samples
All animal procedures were approved by the Committee on Animal Care of Soochow University and were performed in accordance with the approved guidelines. Human venous blood was obtained from healthy donors who read and signed the written informed consent in accordance with the Declaration of Helsinki with permission from the University Ethical Committee of Soochow University.
Statistical analysis
When necessary, the data were analyzed by using Graph Pad Prism 5.0 software and presented as the mean±SD (standard deviation). Statistical significance was determined by two-way ANOVA analysis of variance with the Bonferroni post-test for multiple groups. Student's t test was used to calculate P-values for differences. Differences were considered significant at P<0.05.
Results
SC99 inhibits STAT3 signaling in platelets
To determine STAT3 activation upon treatment of platelet activators, platelets were stimulated with collagen or thrombin at various concentrations or treatment periods. The immunoblotting assay showed that the phosphorylation of STAT3 at Tyr705, an indicator of STAT3 activation, was triggered by both collagen and thrombin. However, with the increase of the concentrations of both collagen and thrombin, the activation level of STAT3 was decreased (Figure 1A). This effect was likely due to the saturation of the receptors on the platelets, but the detailed mechanisms are not currently known. The optimal concentration to activate STAT3 in platelets is 2 μg/mL and 0.02 U/mL for collagen and thrombin, respectively (Figure 1A). At the optimal concentration of collagen and thrombin, the phosphorylation level of STAT3 reached the plateau within 5 to 10 min (Figure 1B). Next, we evaluated the effects of SC99 on STAT3 activation at the optimal concentrations of both collagen and thrombin in 5 min. As shown in Figure 1C, collagen and thrombin markedly increased the phosphorylation level of STAT3, which was inhibited by SC99 in a concentration-dependent manner. In addition, 5 μmol/L SC99 almost completely inhibited STAT3 phosphorylation in platelets stimulated by both collagen and thrombin (Figure 1C).

SC99 inhibits STAT3 activation in human platelets. (A) Platelets (250 μL, 3×108/mL) were stimulated with collagen or thrombin at indicated concentrations for 5 min at 37 °C, and the reaction was stopped by adding RIPA buffer. After heating to 97 °C for 10 min, proteins were fractionated via 10% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed using an immunoblotting assay to evaluate the STAT3 phosphorylation level. (B) Platelets were treated with collagen (2 μg/mL) or thrombin (0.02 U/mL) for the indicated periods. The platelets were then lysed and analyzed using an immunoblotting assay to evaluate the STAT3 phosphorylation level. (C) Starved platelets were pre-treated with SC99 at indicated concentrations for 10 min at 37 °C followed by stimulation with collagen (2 μg/mL) or thrombin (0.02 U/mL) for 5 min. Cell lysates from platelets were then analyzed using an immunoblotting assay to evaluate STAT3 and JAK2 phosphorylation levels. (D) Platelets were treated as described in (C) and were analyzed using an immunoblotting assay to evaluate the phosphorylation levels of p65, p-c-Src, and p-AKT using specific antibodies. β-Actin was used as an internal loading control.
Several important signaling pathways are associated with STAT3 activation, including MAPK/ERK18, PI3K/AKT19, c-Src20, and JAK pathways18. Our previous studies showed that SC09 had no effects on these kinases with the exception of JAK2 in multiple myeloma cells16, we next confirmed the effects of SC99 on these kinases in platelets. As shown in Figure 1C, SC99 significantly suppressed the activation of JAK2 in platelets. In contrast, SC99 had no effects on c-Src or AKT phosphorylation (Figure 1D). Notably, although both STAT3 and p65 are important nuclear transcription factors, SC09 had no effects on p65 (Figure 1D). These results suggested that SC99 inhibits JAK2 and STAT3 activation in platelets.
SC99 inhibits platelet aggregation
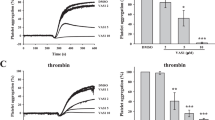
Because STAT3 signals modulate platelet aggregation and the above studies revealed that SC99 inhibited STAT3 activation, we next examined the potential effects of SC99 on platelet aggregation. Washed platelets were incubated with SC99 (0.156, 0.3125, 1.25, or 2.5 μmol/L) or solvent alone for 10 min at 37 °C and then stimulated with collagen (2 μg/mL) or thrombin (0.02 U/mL). As shown in Figure 2, platelet aggregation triggered by both stimulants was inhibited by SC99 in a dose-dependent manner. For example, the maximal aggregation induced by collagen was decreased by 10%, 50% and 70% when pre-incubated with 0.156, 0.3125 and 1.25 μmol/L SC99, respectively (Figure 2A). SC99 also inhibited platelet aggregation induced by thrombin (Figure 2B). In addition, we examined whether low SC99 concentrations were sufficient to disrupt platelet aggregation induced by high doses of collagen or thrombin. Briefly, 0.3125 μmol/L SC99 did not inhibit platelet aggregation triggered by greater than 5 μg/mL collagen (Figure 2E–2G). Similarly, 1.25 μmol/L SC99 failed to inhibit platelet activation by greater than 0.03 U/mL thrombin (Figure 2H–2J). This finding was consistent with results in Figure 2A–2D, and these data suggested that a higher dose of SC99 might be required to achieve the inhibition of platelet aggregation triggered by stronger signals.

SC99 inhibits platelet aggregation. (A, C) Platelets (250 μL, 3×108/mL) were incubated with SC99 at indicated concentrations for 10 min at 37 °C, followed by stimulation with collagen (2 μg/mL) (A) or thrombin (0.02 U/mL) (C) for 1 min, followed by the evaluation of platelet aggregation on a ChronoLog aggregometer. (B and D) represent a statistical report on three independent assays from A and C, respectively. (E–J) Platelets were pre-treated with SC99 (0.312 μmol/L) for 1 min followed by stimulation with increasing collagen (E–G) or thrombin (0.02 U/mL) (H–J) for 5 min. Platelet aggregation was analyzed on a ChronoLog aggregometer. The aggregation curves are the representatives of at least three individual experiments. The platelet aggregation from three experiments was plotted in the bar charts. *P<0.05, **P<0.01 compared with control. Mean±SD. n=3.
SC99 inhibits platelet alpha-granule secretion and inside-out signaling
Platelets are replete with secretory granules that are critical for platelet activation and aggregation. Upon activation, the platelets degranulate and release components from the α-granules (such as vWF and P-selectin) and from the electron dense granules (ADP, serotonin, and thromboxane A2)21. These active substances further enforce platelet aggregation and coagulation. Given that SC99 was able to inhibit platelet aggregation, we assessed whether SC99 could modulate platelet degranulation. To this end, human platelets were pre-incubated with SC99 (5 μmol/L) in the presence of fluorescent PE-conjugated P-selectin antibody (P-selectin/CD62P) and then challenged with thrombin (0.05 U/mL) for 10 min at 37 °C because P-selectin is expressed in the activated platelets. As shown in Figure 3A, based on flow cytometry analysis, thrombin triggered the expression of P-selectin on platelets in the absence of SC99. By contrast, SC99 inhibited thrombin-induced P-selectin expression by 84.3% (P<0.01) (Figure 3A). This experiment thus demonstrated that SC99 inhibited platelet degranulation.

SC99 inhibits platelet alpha-granule secretion and inside-out signaling. (A) Human platelets were pre-treated with SC99 for 5 min in the presence of P-selectin and then challenged with thrombin for 30 min. Platelets were then incubated with PE-CD62P antibody and analyzed on a cytometer. (B) A statistical report on three independent assays from (A). (C) Human platelets were pre-treated with SC99 for 5 min in the presence of FITC-fibrinogen and then challenged with thrombin for 30 min. Platelets were analyzed on a cytometer. (D) A statistical report on three independent assays from (C). **P<0.01 compared with the control and SC99 treatment (n=3).
Most of the contents from the platelet granules further amplify platelet activation, platelet aggregates, and thrombus stabilization via the inside-out signaling pathways, one of which is the integrin αIIbβ3/Fibrinogen signaling. To investigate whether SC99 affected inside-out integrin signaling, we examined the effects of SC99 on integrin αIIbβ3 activation using soluble fibrinogen that binds integrin αIIbβ3. As shown in Figure 3B, thrombin markedly increased the binding of soluble fibrinogen to the platelets, which were diminished by SC99 (up to 75.7% was inhibited, P<0.01). Therefore, these data suggested that SC99 modulated platelet secretion and inside-out signaling.
SC99 modulates outside-in signaling in platelets
Platelet activation and the subsequent inside-out signal cascade further initiates a series of intracellular or outside-in signaling, leading to platelet spreading, granule secretion, stable adhesion, and clot retraction22. The above studies revealed that SC99 exhibited potent inhibition on platelet aggregation and fibrinogen binding, which reflect inside-out signaling. To determine whether SC99 affects outside-in signaling, platelet spreading on immobilized fibrinogen was assessed. As shown in Figure 4, the average surface coverage of vehicle-treated platelets was 19.29±4.83 μm2, which was significantly reduced by SC99 in a dose-dependent manner. At 5 μmol/L, SC99 reduced platelet surface coverage by 78% (4.21±0.79 μm2) compared with the vehicle control. Therefore, SC99 modulates platelet function by inhibiting both inside-out and outside-in signal transduction.

SC99 inhibits the outside-in signaling in human platelets. Platelets were pre-incubated with SC99 at 5 μmol/L for 10 min. Then, platelets were added to fibrinogen-coated coverslips and further incubated for an additional 60 min. The platelet morphology was then analyzed on a fluorescent microscope. (B) The statistical analyses of three independent assays from (A). **P<0.01 compared with the control (n=3).
SC99 slows down clot retraction
Platelet clot retraction is an essential step in thrombus consolidation as a late consequence of integrin outside-in signaling. To investigate whether SC99 interfered with this important outcome, we performed a platelet clot retraction assay by pretreating platelets with SC99 or vehicle. Fibrin clots were induced at 37 °C by the addition of thrombin to platelet suspension, and the subsequent platelet-dependent clot retraction was monitored over a 30-min time period. As shown in Figure 5, the presence of SC99 at 5 μmol/L resulted in marked reduction of clot retraction. The clot sizes with SC99 treatment at 10, 15 and 20 min were approximately two-fold compared with the control.

SC99 delays clot retraction. (A) Platelet-rich plasma was incubated with SC99 for 10 min followed by dispensing 0.25-mL aliquots. Each aliquot was added thrombin to allow clot retraction. Clots were observed at indicated time periods and photographed. (B) The statistical presentation of three independent analysis of the clot retraction at each time point. *P<0.05, **P<0.01 compared with the control (n=3).
SC99 does not induce toxic or apoptotic effects on platelets
To examine whether the inhibitory effect of SC99 on agonist-induced platelet aggregation was due to its potential toxicity to platelets, we performed an AlamarBlue assay. As shown in Figure 6A, there was no marked reduction in the fluorescence intensity in platelets treated with 5 or 10 μmol/L SC99 compared with the vehicle control, indicating that SC99 was not toxic to platelets (Figure 6A). This effect was further evaluated by an Annexin V staining assay in which Annexin V identifies and binds to phosphatidylserine (PS) exposed on the outside of plasma membrane. The results showed that Annexin V-positive platelets were not significantly altered in platelets treated with SC99 (Figure 6B). In combination with the AlamarBlue cell viability assay, we concluded that SC99 was not toxic to platelet viability and confirmed that the inhibitory effect of SC99 on platelet aggregation did not result from apoptotic effects.

SC99 does not induce platelet apoptosis or extend bleeding time. (A) Platelets were treated with SC99 at indicated concentrations for 15 min at 37 °C followed by incubation with AlamarBlue reagent (10 μL) and incubation for an additional 4 h at 37 °C. Live platelets were analyzed on a plate reader with the excitation and emission wavelengths 570 nm and 585 nm, respectively. (B) Platelets were treated with SC99 at indicated concentrations for 15 min at 37 °C followed by Annexin V-FITC and PI staining. Apoptotic platelets were analyzed on a flow cytometer. (C) Ten mice were administered ip with SC99 (2 mg/kg) or vehicle. Then, 45 min later, the distal 5 mm tails were removed. The tails were immediately immersed in warm 0.9% NaCl solution for 10 min. Time to bleeding cessation was recorded. NS: not significant. (D) Mice were administered ip with SC99 (2 mg/kg) or vehicle. Then, 45 min later, blood obtained from inferior vena cava was subjected to platelet isolation. Purified platelets were then stimulated with collagen (2 μg/mL) for 1 min followed by evaluation of platelet aggregation as described previously. Three independent assays were performed.
Given that prolonged bleeding time is an important side effect of anti-platelet drugs, we next examined whether SC99 affected hemostasis while inhibiting platelet activation by comparing the tail-bleeding time. Mice were received SC99 at a dose of 2 mg/kg, followed by soaking the transected tail in physiological buffer and measuring the bleeding time. As shown in Figure 6C, SC99 did not significantly prolong the tail bleeding time (258.7±72.3 s; n=11) compared with the vehicle-treated mice (214.2±66.65 s; n=10) (Figure 6C). Notably, the treatment with SC09 in mice maintained greater than 50% inhibition of platelet aggregation (Figure 6D). Therefore, these studies demonstrated that SC99 inhibited platelet aggregation and was not toxic to platelets.
Discussion
Platelet activation and aggregation is critical in a panel of physiological and pathophysiological processes including hemostasis, thrombosis, wound healing, atherosclerosis, and inflammation. This platelet function must be tightly regulated. Dysregulated thrombus formation causes blockage of blood vessels, leading to ischemia and resulting in thrombotic diseases, such as heart attack and ischemic stroke. Diverse signaling transduction cascades participate in platelet activation and aggregation, such as c-Src, the PI3K/Akt signaling pathway, the NO/cGMP/PKG pathway, and the signaling pathways of MAPK isoforms p38, ERK, and JNK. Recent studies indicate that the STAT3 signaling pathway is also extensively involved in platelet functions14,23. As a nuclear transcription factor, STAT3 is believed to regulate the maturation of megakaryocytes and platelet production23. In mature platelets that lack nuclei, STAT3 modulates platelet activation and aggregation in a nontranscriptional manner14,24. The inhibition of STAT3 signaling leads to platelet inactivation and prevents platelet aggregation and spreading; therefore, STAT3 could be a novel therapeutic target for thrombotic diseases. In the present study, we demonstrated that SC99 is a novel STAT3 inhibitor that could be used as an anti-platelet agent to treat thrombotic diseases.
Consistent with our expectation, several lines of experimental evidence confirmed that STAT3 signaling regulates collagen and thrombin-induced platelet activation and aggregation, which could be inhibited by SC99 in a concentration- and time-course-dependent manner. Notably, SC99 was not toxic to platelet viability, and it did not significantly prolong the bleeding time. Thus, the inhibitory effect of SC99 on platelet aggregation did not result from apoptotic effects. In contrast, SC99 specifically inhibited the phosphorylation of STAT3 but not Akt, p65 and c-Src, which is consistent with our findings in other cells16.
Platelet receptors receive stimulations from adhesive proteins exposed in an injured vessel wall and soluble platelet agonists, transmit the signals via their platelet activation pathway, and ultimately induce the inside-out signaling process that leads to integrin activation. STAT3 signaling is one of the important pathways to mediate receptor signals and promote αIIbβ3-talin interaction and integrin activation25. SC99 inhibited platelet inside-out signaling as it attenuates fibrinogen binding to integrin αIIbβ3 by the thrombin receptor ligand. This integrin signaling is critically important in stabilizing platelet adhesion, spreading, and clot retraction26,27. This integrin signal is associated with MAPK (including p38 and ERK) and JNK activation28. However, our study did not indicate that SC99 inhibited ERK and p38 phosphorylation stimulated by collagen and thrombin in platelets. In contrast, we found that SC99 inhibited STAT3 phosphorylation. STAT3 activation in collagen-activated platelets involves Syk-PLCγ2 signaling15. Moreover, SC99 inhibited platelet clot retraction, indicating that SC99 probably acts as an effective modulator for platelet contractile signaling and platelet-dependent clot retraction. However, it is unclear how SC99 inhibits the STAT3 signaling pathway, which should be further investigated14.
Conclusions
The STAT3 pathway is involved in collagen- and thrombin-induced platelet activation. By inhibiting STAT3 signaling, the novel compound SC99 displays potent anti-platelet activity but has no marked effects on bleeding and platelet activity. Our findings suggest that STAT3 signaling may represent a novel therapeutic target for preventing and treating thromboembolic diseases, and SC99 could be developed as a promising agent for the treatment of these diseases.
Author contribution
Yan KONG and Xin-liang MAO designed the study, analyzed the data, and wrote the manuscript; Zhuan XU, Yu-jia XU, Ya-nan HAO, Li-jie REN, Xin XU, Zu-bin ZHANG, and Bi-yin CAO conducted the experiments; Ke-sheng DAI, Li ZHU, and Qi FANG performed the data analysis. All authors read and approved the final manuscript.
References
Nieswandt B, Watson SP . Platelet-collagen interaction: is GPVI the central receptor? Blood 2003; 102: 449–61.
Ozaki Y, Asazuma N, Suzuki-Inoue K, Berndt MC . Platelet GPIb-IX-V-dependent signaling. J Thromb Haemost 2005; 3: 1745–51.
Wu Y, Asazuma N, Satoh K, Yatomi Y, Takafuta T, Berndt MC, et al. Interaction between von Willebrand factor and glycoprotein Ib activates Src kinase in human platelets: role of phosphoinositide 3-kinase. Blood 2003; 101: 3469–76.
Marshall SJ, Senis YA, Auger JM, Feil R, Hofmann F, Salmon G, et al. GPIb-dependent platelet activation is dependent on Src kinases but not MAP kinase or cGMP-dependent kinase. Blood 2004; 103: 2601–9.
Jurk K, Kehrel BE . Platelets: physiology and biochemistry. Semin Thromb Hemost 2005; 31: 381–92.
Chen K, Rondina MT, Weyrich AS . A sticky story for signal transducer and activator of transcription 3 in platelets. Circulation 2013; 127: 421–3.
Shih CH, Chiang TB, Wang WJ . A critical role for the regulation of Syk from agglutination to aggregation in human platelets. Biochem Biophys Res Commun 2014; 443: 580–5.
Metharom P, Berndt MC, Baker RI, Andrews RK . Current state and novel approaches of antiplatelet therapy. Arterioscler Thromb Vasc Biol 2015; 35: 1327–38.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 2015; 131: e29–322.
Greenlund AC, Morales MO, Viviano BL, Yan H, Krolewski J, Schreiber RD . Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity 1995; 2: 677–87.
Reich NC, Liu L . Tracking STAT nuclear traffic. Nat Rev Immunol 2006; 6: 602–12.
Kaushansky K . Thrombopoietin: the primary regulator of platelet production. Blood 1995; 86: 419–31.
Drachman JG, Sabath DF, Fox NE, Kaushansky K . Thrombopoietin signal transduction in purified murine megakaryocytes. Blood 1997; 89: 483–92.
Zhou Z, Gushiken FC, Bolgiano D, Salsbery BJ, Aghakasiri N, Jing N, et al. Signal transducer and activator of transcription 3 (STAT3) regulates collagen-induced platelet aggregation independently of its transcription factor activity. Circulation 2013; 127: 476–85.
Lu WJ, Lin KC, Huang SY, Thomas PA, Wu YH, Wu HC, et al. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb Res 2014; 133: 1088–96.
Zhang Z, Mao H, Du X, Zhu J, Xu Y, Wang S, et al. A novel small molecule agent displays potent anti-myeloma activity by inhibiting the JAK2-STAT3 signaling pathway. Oncotarget 2016; 7: 9296–308.
Yi W, Li Q, Shen J, Ren L, Liu X, Wang Q, et al. Modulation of platelet activation and thrombus formation using a pan-PI3K inhibitor S14161. PLoS One 2014; 9: e102394.
Aznar S, Valeron PF, del Rincon SV, Perez LF, Perona R, Lacal JC . Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol Biol Cell 2001; 12: 3282–94.
Chew GS, Myers S, Shu-Chien AC, Muhammad TS . Interleukin-6 inhibition of peroxisome proliferator-activated receptor alpha expression is mediated by JAK2- and PI3K-induced STAT1/3 in HepG2 hepatocyte cells. Mol Cell Biochem 2014; 388: 25–37.
Silva CM . Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004; 23: 8017–23.
Polasek J . Procoagulant potential of platelet alpha granules. Platelets 2004; 15: 403–7.
Shen B, Delaney MK, Du X . Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol 2012; 24: 600–6.
Oda A, Miyakawa Y, Druker BJ, Ishida A, Ozaki K, Ohashi H, et al. Crkl is constitutively tyrosine phosphorylated in platelets from chronic myelogenous leukemia patients and inducibly phosphorylated in normal platelets stimulated by thrombopoietin. Blood 1996; 88: 4304–13.
Miyakawa Y, Oda A, Druker BJ, Miyazaki H, Handa M, Ohashi H, et al. Thrombopoietin induces tyrosine phosphorylation of Stat3 and Stat5 in human blood platelets. Blood 1996; 87: 439–46.
Moore DT, Nygren P, Jo H, Boesze-Battaglia K, Bennett JS, DeGrado WF . Affinity of talin-1 for the beta3-integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc Natl Acad Sci U S A 2012; 109: 793–8.
Li Z, Delaney MK, O'Brien KA, Du X . Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 2010; 30: 2341–9.
Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF . Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest 2004; 113: 441–50.
Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X . Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009; 113: 893–901.
Acknowledgements
This work was partly supported by the Natural Science Foundation of Jiangsu Province (BK20160347 and BE2014630), the National Natural Science Foundation of China (81272632 and 81301906), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-psycho-diseases (BM2013003).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Xu, Z., Xu, Yj., Hao, Yn. et al. A novel STAT3 inhibitor negatively modulates platelet activation and aggregation. Acta Pharmacol Sin 38, 651–659 (2017). https://doi.org/10.1038/aps.2016.155
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.155
Keywords
This article is cited by
-
Podocarpusflavone alleviated renal fibrosis in obstructive nephropathy by inhibiting Fyn/Stat3 signaling pathway
Journal of Natural Medicines (2023)
-
Platelet-Neutrophil Interactions and Thrombo-inflammatory Complications in Type 2 Diabetes Mellitus
Current Pathobiology Reports (2022)
