
Abstract
Hepatic ischemia-reperfusion (I/R) injury is a common clinical impairment that occurs in many circumstances and leads to poor prognosis. Both apoptosis and autophagy have been shown to contribute to cell death in hepatic I/R injury. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is one of the best-studied anti-inflammatory prostaglandins, which has been verified to exert anti-inflammatory and cell-protective functions in various types of cells and animal models. In this study we explored the effects of 15d-PGJ2 on both apoptosis and autophagy in mouse hepatic I/R injury and its possible mechanisms. A model of segmental (70%) hepatic warm ischemia was established in Balb/c mice, and the pathological changes in serum and liver tissues were detected at 6, 12, and 24 h post-surgery, while 15d-PGJ2 (2.5, 7.5, 15 μg, iv) was administered 30 min prior the surgery. Pretreatment with 15d-PGJ2 (7.5, 15 μg) significantly ameliorated I/R-induced hepatic injury evidenced by dose-dependent reduction of serum ALT and AST levels as well as alleviated tissue damages. 15d-PGJ2 pretreatment significantly decreased the serum TNF-α and IL-1β levels and the hepatic expression of F4/80, a major biomarker of macrophages. 15d-PGJ2 pretreatment upregulated the Bcl-2/Bax ratio, thus reducing the number of apoptotic cells in the livers. 15d-PGJ2 pretreatment considerably suppressed the expression of Beclin-1 and LC3, thus decreasing the number of autophagosomes in the livers. Furthermore, 15d-PGJ2 pretreatment activated Nrf2 and inhibited a ROS/HIF1α/BNIP3 pathway in the livers. Pretreatment with the PPARγ receptor blocker GW9662 (2 μg, ip) partly reversed the protective effects of 15d-PGJ2 on hepatic I/R injury. In conclusion, our results confirm the protective effect of 15d-PGJ2 on hepatic I/R injury, an effect that may rely on a reduction in the activation of Kupffer cells and on activation of the Nrf2 pathway, which lead to inhibition of ROS generation, apoptosis, and autophagy.
Similar content being viewed by others

Introduction
Hepatic ischemia-reperfusion (I/R) injury is a common clinical impairment that occurs in many circumstances, such as liver transplantation, liver hemorrhagic shock, trauma, and cancer1,2. The hepatic I/R process is characterized by interruption of liver blood flow and blockage of oxygen supply, followed by reestablishment of blood flow and oxygen supply, and the consequent disruption of liver cellular metabolism and redox status, which causes impairment of liver tissue function1,2. Hepatic I/R injury may cause organ dysfunction or even primary non-function after trauma or poor graft function after liver transplantation, which leads to poor prognosis5,6.
The complicated mechanisms of hepatic I/R injury have been widely studied7. The activation of Kupffer cells (KCs) is thought to initiate hepatic I/R injury, and it is followed by the release of a series of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α) and interleukin (IL)-1β, the expression of cell adhesion factors, and the production of reactive oxygen species (ROS)8. TNF-α is a major effector cytokine in hepatocellular and endothelial injuries that induces leukocyte chemotaxis, activates neutrophils, and generates free radicals, as well as induces mitochondrial toxicity and apoptotic cell death through activation of caspases9. In addition, TNF-α can also lead to the release of IL-1β by mediating the generation of inflammasomes10. ROS are produced by KCs in the beginning of hepatic I/R injury, and they are also generated when liver cells are deprived of nutrients or have mitochondrial injury11. ROS accumulation occurs downstream of many detrimental pathways. Furthermore, ROS in the radical form are also effector molecules that cause severe damage to DNA, RNA, and proteins directly12,13.
In hepatic I/R injury, a dead liver cell is usually present in a necrotic form, with a rapid swelling of cells and cell organelles, accompanied by abnormal membrane stretching and eventual cellular rupture14,15. In addition to necrosis, malfunctioning programmed cell death (PCD) is also observed in the hepatic I/R process16. Apoptosis, known as type 1 PCD, is characterized by a series of biochemical and morphological changes, including caspase activation, chromosomal DNA cleavage, nuclear condensation and fragmentation, and cell shrinkage, as well as loss of adhesion to neighboring cells or the extracellular matrix17,18. Autophagy is a catabolic process crucial for maintaining cell homeostasis and is defined as type 2 PCD. Autophagy is a process involving the formation of double-membrane-bound structures called autophagosomes, which surround cytoplasmic macromolecules or organelles, and fuse with lysosomes to form autolysosomes, wherein cellular components are degraded19,20. Both apoptosis and autophagy are two major mechanisms involved in responding to extracellular or intracellular stress caused by inflammatory cytokines such as IL-1β and TNF-α and other stress signals such as ROS21,22,23.
Recent studies have suggested that nuclear factor-erythroid 2-related factor 2 (Nrf2) may be a key transcription factor involved in protecting cells from oxidative stress. Nrf2 is retained in the cytoplasm by binding to its inhibitor, Kelch-like ECH-associated protein 1 (Keap1), under physiological conditions24. Oxidative stress can dissociate Nrf2 from Keap1 and induce the nuclear translocation of Nrf2 and a consequent upregulation in the expression of several antioxidant genes and detoxification enzymes, such as glutathione-S-transferases (GSTs), reduced nicotinamide adenine dinucleotide phosphate (NADPH), quinine oxidoreductase 1 (NQO1), glutamate cysteine ligase (GCL), and heme oxygenase 1 (HO-1), which in turn eliminate ROS and reduce oxidative stress25,26. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), a dehydration product of prostaglandin (PG) D2, is one of the best-studied anti-inflammatory prostaglandins and has been verified to exert anti-inflammatory and cell-protective functions in various types of cells and animal models27,28. Previous studies have demonstrated the protective effects of 15d-PGJ2 on I/R injury in the brain, myocardium, and kidney, and linked the protective effects to the activation of Nrf229,30.
In the present study, the protective effect of 15d-PGJ2 on hepatic I/R injury has been confirmed, and the effects may rely on a reduction of KC activation and on the activation of the Nrf2 pathway, thereby inhibiting ROS generation, apoptosis, and autophagy.
Materials and methods
Reagents
15d-PGJ2 and PPARγ receptor blocker GW9662 were purchased from Sigma-Aldrich (St Louis, MO, USA). Antibodies used in this study included those directly against TNF-α (Abcam, USA), IL-1β (Abcam, USA), Bcl-2 (Cell Signal Technology, USA), Bax (Proteintech, China), Nrf2 (CST, USA), Beclin-1 (Proteintech, China), microtubule-associated protein 1A/1B light chain 3 (LC3) (CST, USA), hypoxia-inducible factor 1α (HIF1α) (Abcam, USA), peroxisome proliferator-activated receptor γ (PPARγ) (CST, USA) and Bcl-2/adenovirus E1B 1 kDa protein-interacting protein 3 (BNIP3) (Abclonal, China). The alanine aminotransferase (ALT) and aspartate aminotransferase (AST) microplate test kits were purchased from Nanjing Jiancheng Bioengineering Institute (Jiancheng Biotech, China).
Animal preparation
A total of 105 male Balb/c mice (7 weeks old, 22±2 g) were purchased from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, China). The mice were raised in a clean room maintained at 24±2 °C under a 12 h:12 h light:dark cycle, with free access to food and water. All animal experiments were approved by the Animal Care and Use Committee of Shanghai Tongji University. The mice were divided randomly into five groups: the control group (three for each time point), I/R model (six for each time point), I/R model+2.5 μg 15d-PGJ2 (six for each time point), I/R model+7.5 μg 15d-PGJ2 (six for each time point), and I/R model+15 μg 15d-PGJ2 (six for each time point). 15d-PGJ2 (dissolved in methyl acetate) was diluted with pyrogen-free normal saline solution and injected via the tail vein 30 min before I/R. GW9662 was intraperitoneally injected at 2 μg 30 min before the administration of 15 μg 15d-PGJ2.
I/R model establishment
A model of segmental (70%) hepatic warm ischemia was used in this study as described in a previous study2. In brief, after fasting for 24 h, mice were anesthetized with 1.25% Nembutal (St Louis, MO, USA) and placed on a sterile experimental table. A midline laparotomy was then performed on all mice. All of the structures in the portal triad (hepatic artery, portal vein, and bile duct) connected to the left and median liver lobes were occluded with metal microvascular clamps. After 60 min, the clamps were loosened to reestablish the blood flow and achieve reperfusion. After reperfusion, the abdominal cavity was closed with surgical sutures, and the mice were placed in a warm environment until they woke up.
Sample collection
In each group, three or six mice were randomly sacrificed at the designated time points: 6, 12, and 24 h after I/R. Tissues of the median and left liver lobes (stored at -80 °C) and orbital blood (stored at 4 °C) were collected.
Serum aminotransferase and cytokine assessment
Serum samples were collected by centrifuging blood samples at 2000×g for 10 min. Serum levels of ALT and AST were measured with ALT and AST microplate test kits according to the manufacturer's instructions. Serum levels of the inflammatory cytokines TNF-α and IL-1β were measured using ELISA kits purchased from eBioscience, USA.
Histopathology
The appropriate size of the liver tissue in each mouse was removed and fixed in 4% paraformaldehyde for 24 h. Liver samples were embedded in paraffin after fixation and cut into 5-μm-thick sections, which were then stained with hematoxylin and eosin (H&E) to observe the pathological structures of the liver by microscopy.
Immunohistochemistry
Liver tissues were prepared in paraffin-embedded sections, dewaxed in xylene, and dehydrated in alcohol. Antigen retrieval was achieved by citrate buffer and incubation in a 95 °C water bath for 20 min. To block endogenous peroxidase, the sections were incubated with 3% hydrogen peroxide for 10 min at 37 °C. Non-specific proteins were blocked with 5% bovine serum albumin (BSA) for 30 min. Liver sections were then incubated overnight with rabbit anti-mouse Nrf2 (1:100), anti-Beclin-1 (1:50), anti-LC3A/B (1:50), anti-HIF1α (1:50), or anti-BNIP3 (1:50) antibody at 4°C, and were then washed with phosphate buffer saline (PBS) and treated immediately with the secondary antibody (goat anti-rabbit) (Epitomics, CA, USA) for another 30 min on d 2. The analysis of antibody binding was performed using a diaminobenzidine (DAB) kit, and sections were counterstained with hematoxylin. Slices were then observed under a light microscope, and five fields for each liver slice were randomly selected for quantitation.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
A TUNEL staining kit was used according to the manufacturer's instructions (Beyotime Biotechnology, Shanghai, China). The liver slices were processed with dewaxing, dehydration, and rehydration. Then, liver slices were stained with the DAB kit and counterstained with hematoxylin. The brown nuclear cells were regarded as positive cells and counted. Three different fields of vision were randomly selected for each slide, and the average percentage of apoptotic cells to total cells was calculated.
Western blot analysis
Liver tissues were rapidly lysed with a mixture of radioimmunoprecipitation assay (RIPA) lysis buffer and protease inhibitors. Tissue lysates were collected and either processed with the nuclear extract kit (Pierce, USA) to isolate the nuclear proteins following the manufacturer's protocol or subjected to total protein extraction procedures. The protein concentration was determined with the bicinchoninic acid (BCA) method, and the proteins were mixed with 5× sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer to be boiled. Total proteins (120 μg) were subjected to SDS-PAGE and blotted onto a polyvinylidene difluoride (PVDF) membrane (Millipore Corp, Billerica, MA, USA). Non-specific binding was blocked with 5% BSA (dissolved in PBS) for 2 h, and then the proteins were incubated overnight at 4°C with the following antibodies diluted in 5% BSA: rabbit anti-mouse TNF-α (1:1000), anti-IL-1β (1:2500), anti-Bcl-2 (1:1000), anti-Bax (1:1000), anti-Nrf2 (1:500), anti-Beclin-1 (1:1000), anti-LC3A/B (1:500), anti-HIF1α (1:500), anti-PPARγ (1:1000), and anti-BNIP3 (1:500). All membranes were washed with PBS with 0.1% Tween 20 (PBST) three times and then incubated with secondary antibodies for 1 h at 37 °C. Finally, membranes were again washed with PBST three times for 5 min each time, and proteins were detected by fluorescence by using the Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Real-time reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was extracted from frozen liver tissue using TRIzol reagent (Tiangen Biotech, China) as described by the manufacturer. cDNA was synthesized using the RT Kit (TaKaRa Biotechnology, China). Gene expression was detected with cDNA SYBR Premix EX Taq (TaKaRa Biotechnology, China). Finally, the results were measured using a 7900HT fast real-time PCR system (ABI, CA, USA). Primer sequences were as follows: TNF-α, forward 5′-CAGGCGGTGCCTATGTCTC-3′, reverse 5′-CGATCACCCCGAAGTTCAGTAG-3′; IL-1β, forward 5′-CGATCGCGCAGGGGCTGGGCGG-3′, reverse 5′-AGGAACTGACGGTACTGATGGA-3′; LC3, forward 5′-GACCGCTGTAAGGAGGTGC-3′, reverse 5′-AGAAGCCGAAGGTTTCTTGGG-3′; Beclin-1, forward 5′-ATGGAGGGGTCTAAGGCGTC-3′, reverse 5′-TGGGCTGTGGTAAGTAATGGA-3′; Bax, forward 5′-AGACAGGGGCCTTTTTGCTAC-3′, reverse 5′-AATTCGCCGGAGACACTCG-3′; β-actin, forward 5′-GGCTGTATTCCCCTCCATCG-3′, reverse 5′-CCAGTTGGTAACAATGCCATGT-3′; Bcl-2, forward 5′-GCTACCGTCGTGACTTCGC-3′, reverse 5′-CCCCACCGAACTCAAAGAAGG-3′; HIF1α, forward 5′-ACCTTCATCGGAAACTCCAAAG-3′, reverse 5′-CTGTTAGGCTGGGAAAAGTTAGG-3′; BNIP3, forward 5′-CTGGGTAGAACTGCACTTCAG-3′, reverse 5′-GGAGCTACTTCGTCCAGATTCAT-3′.
ROS assay assessment
Fresh liver tissues of each mouse were fixed in 4% paraformaldehyde on ice for 1 h. The fixed tissues were then washed with PBS and dehydrated in 30% sucrose at 4 °C overnight. Then, the tissues were infiltrated with OCT (Sakura, USA) for 2 h and preserved at -80 °C. Sections (5 μm) cut with a freezing microtome were dried at room temperature for 5 min and then washed three times with PBS for 5 min. Avoiding light, sections were incubated with ROS Fluorescent Probe-DHE (vigorous, Beijing, China) (50 μmol/L, diluted by PBS) for 75 min and washed with PBS as before. The prepared sections were finally sealed with quenching agent and observed under a fluorescence microscope.
Transmission electron microscopy
Liver tissues were preserved with 2 mL of 2.5% glutaraldehyde in PBS and fixed in 1% OsO4. Livers were sectioned and photographed by transmission electron microscopy (JEOL, JEM 1230, Japan) at 80 or 60 kV onto an electron microscope film (Kodak, ESTAR thick base, NY, USA), and then images were printed onto photographic paper.
Statistical analysis
All results are presented in the format of mean±standard deviation (SD). The results of ALT, AST, qRT-PCR, ELISA, Western blot, and immunohistochemistry were analyzed using Student's t-test. In all comparisons, P<0.05 was considered statistically significant. All statistical analyses were performed with SPSS 20.0 for Windows (IBM, Armonk, NY, USA).
Results
15d-PGJ2 pretreatment ameliorates liver injury induced by hepatic I/R
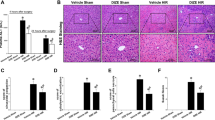
In the hepatic I/R process, damaged and ruptured liver cells release aminotransferases such as ALT and AST into the bloodstream. Elevated levels of serum ALT and AST reflect the degree of liver and hepatic cell damage. As shown in Figure 1A, the levels of serum ALT and AST from each group of mice were measured. After I/R administration, the level of aminotransferases increased significantly in the model group when compared to the control group. The value rose at 6 h, peaked at 12 h, and was maintained at a relatively high level until 24 h. The 15d-PGJ2-treated groups showed protective effects according to the dose: the 2.5 μg dose showed no differences from the I/R model at all three time points; the 7.5 μg dose had a reduced aminotransferase levels at 6 and 12 h (P<0.05) but no significant differences at 24 h from the I/R model; the 15 μg dose showed a protective effect at all three time points (P<0.05). The morphological changes shown by H&E staining in the 15d-PGJ2 pretreatment groups also indicated the different efficiencies among the doses. After I/R administration, necrotic hepatocytes were characterized by condensation of chromatin, swelling of organelles, and consequently karyolysis and rupture of the whole cell. The staining of necrotic hepatocyte nuclei faded, and disintegrated necrotic cells and stromata integrated into a blur of unstructured eosinophilic substance that presented as a lighter color in H&E staining, which could be clearly observed in the I/R model group when compared to the ordered structures of the morphologically normal cells in the control group. The pathological features were clearly reduced in the I/R model+15 μg 15d-PGJ2 group, as shown in Figure 1B. It can be concluded that the administration of 2.5 μg 15d-PGJ2 had no effect, and it was not tested in the subsequent experiments.

15d-PGJ2 pretreatment ameliorates hepatic I/R injury (A) the index of plasma ALT and AST levels at 6, 12, and 24 h after I/R administration in mice, and effects of 15d-PGJ2 treatment groups at the same time. 15d-PGJ2 groups showed varying degrees of protective effects according to its doses: the 2.5 μg dose showed no differences with I/R model among all three time points; the 7.5 μg dose had a reduced aminotransferase level at 6 and 12 h (P<0.05) but no statistical differences at 24 h with I/R model; the 15 μg dose, obviously, showed a protective effect in all three time points (P<0.05). Data are expressed as mean±SD. n=6. *P<0.05 for negative control (NC) group vs I/R. #P<0.05 for I/R vs I/R+7.5 or 15 μg 15d-PGJ2. (B) Hematoxylin and eosin staining of liver sections. Necrosis can mainly be observed in I/R model groups and 2.5 μg 15d-PGJ2 groups, areas expanded over time. Black bar for 200 μm. Black arrow for necrotic area.
15d-PGJ2 reduces F4/80 expression and TNF-α and IL-1β production
As previously mentioned, KCs are the most important inflammatory cells in the initial phase of hepatic I/R injury. Pro-inflammatory cytokines such as TNF-α and IL-1β are released by activated KCs, and induce hepatocellular and endothelial injuries, leading to cell necrosis. Serum levels of TNF-α and IL-1β were first measured, and the results showed that both cytokines rose from 6 h, peaked rapidly at 12 h, and maintained a high level at 24 h (Figure 2A). Transcription levels were also detected by qRT-PCR and protein expression by Western blot. The administration of both 7.5 and 15 μg 15d-PGJ2 reduced the levels of pro-inflammatory cytokines at all three time points (Figure 2B, 2C). The immunohistochemistry results showed different expression levels of F4/80, a major biomarker of macrophages, between the control and I/R model groups, indicating an activation of macrophages in response to I/R injury. Analogously, 15d-PGJ2 exhibited a weakened effect on activation of macrophages, as indicated by reduced expression of F4/80-positive cells (Figure 2D).

15d-PGJ2 reduces F4/80 expression and TNF-α and IL-1β productions. (A) The index of plasma TNF-α and IL-1β levels at 6, 12, and 24 h after I/R administration, and effects of 15d-PGJ2 treatment groups at the same time. Data are expressed as mean±SD. n=6. *P<0.05 for NC vs I/R, #P<0.05 for I/R vs I/R+15d-PGJ2. (B) The mRNA expression of TNF-α and IL-1β in NC, I/R and I/R+15d-PGJ2 group were evaluated by Real time PCR. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. Both cytokines rose from 6 h, and peaked rapidly at 12 h and maintained a high level at 24 h. (C) The Western blot analysis of TNF-α and IL-1β. The results of Western blot were analyzed with Quantity One. n=3. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.

The different expression of F4/80 was evaluated by immunohistochemistry at 6 h. The F4/80 positive cells were dyed brown and presented typical form among the hepatocytes. The F4/80 positive cells and total cells were counted and the ratio was calculated in 6 random fields with Image-pro Plus 6.0. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.
15d-PGJ2 reduces the Bax/Bcl-2 ratio and number of apoptotic cells
In addition to necrosis, apoptosis also results in cell death in hepatic I/R injury, thus contributing to hepatic dysfunction. Therefore, to explore the potential protective mechanism of 15d-PGJ2 against hepatic I/R injury, changes in Bcl-2 and Bax at the cDNA and protein levels were measured. The expression of Bcl-2 and Bax cDNA was detected with qRT-PCR, as shown in Figure 3A. It was clear that 15d-PGJ2 pretreatment significantly reduced expression of Bax and simultaneously increased expression of Bcl-2 at the three time points. 15d-PGJ2 also reduced the expression of Bax at the protein level at all three time points, and the expression of Bcl-2 increased mainly at 6 and 12 h with 15d-PGJ2 treatment (Figure 3B). In addition, a similar result was observed for the immunohistochemistry at 8 h (Figure 3C). The apoptotic cells were detected by TUNEL staining. As shown in Figure 3D, TUNEL-positive cells were observed in the I/R model group and their numbers were significantly decreased in 15d-PGJ2 pretreatment groups.

15d-PGJ2 reduces Bax/Bcl-2 ratio and amount of apoptotic cells. (A) The mRNA expression of Bcl-2 and Bax in NC, I/R and I/R+15d-PGJ2 groups were evaluated by Real time PCR. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. Although cDNA level of both Bcl-2 and Bax increased in the I/R model group, 15d-PGJ2 treatment groups showed a more elevated Bcl-2/Bax ratio. (B) The Western blot analysis of Bcl-2 and Bax indicate an inhibition of apoptosis effect by 15d-PGJ2. (C) TUNEL assay in I/R and I/R+15d-PGJ2 groups showed quantitative differences in apoptotic cells. The differend expression of Bcl-2 and Bax were also evaluated by immunohistochemistry after 6 h after I/R administration. (D) TUNEL positive cells and total cells were counted and the ratio was calculated in 6 random fields with Image-pro Plus 6.0. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. The expression of Bcl-2 and Bax in immunohistochemistry were measured by the relative IOD (integrated optical density) with negative control groups respectively. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.
15d-PGJ2 inhibits the expression of Beclin-1 and LC3 and decreases the number of autophagosomes
Beclin-1 and LC3 play pivotal roles in the autophagy process: Beclin-1 integrates upstream signals by combining with other autophagy regulator kinases, while LC3 makes up the membrane of autophagosomes. In the present study, both the cDNA and protein levels of Beclin-1 and LC3 were measured. Figures 4A and 4B show that transcription of Beclin-1 and LC3 was increased from 6 to 24 h in I/R model groups compared with control groups. The results of the Western blot also indicated improved autophagy levels with higher protein expression of Beclin-1 and LC3II. Additionally, electron microscopy was used to observe the ultrastructures of hepatic cells. Significant ultrastructural morphological changes were found in the I/R model group, such as mitochondrial swelling and crest damage and increased numbers of lysosomes and autophagosomes. However, with 15d-PGJ2 pretreatment, liver nuclear chromatin was more homogeneous and the structural integrity was maintained, with fewer lysosomes and autophagosomes (Figure 4D).

15d-PGJ2 ameliorates autophagy in I/R-induced acute liver injury. (A) The qRT-PCR analysis of Beclin-1 and LC3 at 6, 12, and 24 h after I/R administration in mice, and effects of 15d-PGJ2 treatment groups at the same time. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. (B) The Western blot results were analyzed using Quantity One. n=3. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.

(C) The different expression of Beclin-1 and LC3 evaluated by immunohistochemistry and analyzed the relative IOD with negative control groups, respectively. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. (D) The transmission electron microscopy (TEM) was used as a direct observation of the autophagosomes formation in I/R and I/R+15d-PGJ2 groups at 6 h. Red arrow for autophagosomes or autophagolysosomes.
15d-PGJ2 induces nuclear translocation of Nrf2, inhibits expression and translocation of HIF1α, and reduces ROS levels
The cell nucleus was extracted to detect protein expression of Nrf2 and HIF1α. The results of the Western blot showed that the nuclear expression of Nrf2 increased in the I/R model group, whereas it was greatly enhanced in the 15d-PGJ2 treatment groups. A similar trend was observed in the cDNA levels and immunohistochemistry results (Figure 5A, 5C). HIF1α is a major factor involved in the response to hypoxia. HIF1α is stable under hypoxic conditions and can also be induced by ROS. One of the major target genes is BNIP3, which can combine with Bcl-2, separating it from Beclin-1 and thus activating autophagy. Increased HIF1α and BNIP3 cDNA and protein levels were detected in the I/R model group. The results of immunohistochemistry also showed differential expression of HIF1α nuclear translocation between the I/R model and control groups. On the contrary, 15d-PGJ2 reduced the nuclear expression and transcription levels of HIF1α, which was confirmed by immunohistochemistry, and is shown in Figure 5B and 5C. Probe-DHE, which emits red fluorescence after oxidation by ROS, was used to observe ROS production in the liver tissue of all groups. Significantly increased red puncta indicated a significant increase in ROS in the I/R model group compared with the control group. Similarly, 15d-PGJ2 decreased ROS production, as shown in Figure 5C.

15d-PGJ2 induces a nuclear translocation of Nrf2, inhibits expression and translocation of HIF1α and reduces BNIP3 and ROS levels. (A) The cDNA levels of HIF1α and BNIP3 were detected by q-RTPCR, 15d-PGJ2 showed an inhibition effect on HIF1α and BNIP3 transcription at all three time points besides 24 h with a dose of 7.5 μg. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. (B) The nucleus expression of Nrf2 increased in the I/R model group, whereas greatly enhanced in the 15d-PGJ2 treatment groups, detected by Western blot. The increased HIF1α and BNIP3 at protein levels were also detected elevated in the I/R model group and declined in the presence of 15d-PGJ2. n=3. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.

(C) The same trend also appeared in immunohistochemical examination. The DAB-positive nucleus refers to the nuclear translocation of Nrf2 or HIF1α, counted with Image-pro Plus 6.0. n=6. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.
The protective effects of 15d-PGJ2 on hepatic I/R injury are associated with PPARγ, which can be partly reversed by the PPARγ receptor blocker GW9662
To investigate whether the protective effects of 15d-PGJ2 are associated with PPARγ in this model, we further tested the serum levels of aminotransferase and cytokines after using the PPARγ receptor blocker GW9662 in a separate group. GW9662 (2 μg) was intraperitoneally injected 30 min before the administration of 15d-PGJ2 in this group. Significant increases in the serum levels of ALT and AST were observed, while no significant differences between the serum and protein expression of IL-1β and TNF-α were found in the presence of GW9662 (Figure 6A and 6B). However, 15d-PGJ2 and GW9662 co-treatment still showed a protective effect compared with I/R group (Figure 6A). Moreover, the expression of nuclear PPARγ was slightly increased at 6 h after I/R administration, which could be further increased by 15d-PGJ2 pretreatment, as shown in Figure 6C. Therefore, we suggest that PPARγ was partially involved on in the protective effect of 15d-PGJ2, although it may not play a key role in the reduction of pro-inflammatory cytokines by 15d-PGJ2 pretreatment, as in our model.

The protective effects of 15d-PGJ2 are associated with PPARγ. (A) The index of plasma ALT and AST levels at 6 h after I/R administration in mice, and effects of 15 μg 15d-PGJ2 treatment group with or without the PPARγ receptor blocker GW9662 at the same time. In the presence of GW9662, despite a significant decline could been observed (*P<0.05), 15d-PGJ2 still showed a protect effect compared with I/R group (*P<0.05). (B) The index of plasma IL-1β and TNF-α levels at 6 h after I/R administration, and effects of 15 μg 15d-PGJ2 treatment group with or without the PPARγ receptor blocker GW9662 at the same time. The pro-inflammatory cytokines like TNF-α and IL-1β reduced by 15d-PGJ2 is PPARγ-independent and cannot been blocked by GW9662 (NSP>0.05). Data are expressed as mean±SD. n=6. +P<0.05 for I/R groups vs I/R+15d-PGJ2+GW9662. @P<0.05 for I/R+15d-PGJ2 vs I/R+15d-PGJ2+GW9662. (C) The nucleus expression of PPARγ increased in the I/R model group, whereas greatly enhanced in the 15 μg 15d-PGJ2 treatment groups and restored by GW9662, detected by Western blot. The results of Western blot analyze of TNF-α and IL-1β are same with ELISA. The results of Western blot were analyzed with Quantity One. n=3. *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2. NSP>0.05, $P<0.05 I/R+15d-PGJ2+GW9662 vs I/R+15d-PGJ2.
Discussion
Hepatic I/R injury is initiated by a series of alterations of cellular biochemistry after blood and oxygen supplies are broken down. A dysfunction of the adenosine triphosphate-driven 'sodium pump' in settings of hypoxia and reperfusion leads to a disruption of the intercellular ionic balance across the cell membrane, which is accompanied by cell swelling and activation of phospholipases, proteases, and endonucleases, thus promoting structural and membrane loss and eventually causing cell damage and death31,32. Injured and dead liver cells stimulate and activate KCs, and initiate inflammatory responses33. The activation of KCs leads to the release and accumulation of TNF-α and IL-1β. TNF-α induces hepatocellular and endothelial injuries and chemotaxis and activation of polymorphonuclear neutrophils and leukocytes, as well as ROS production by KCs9. TNF-α also leads to a release of IL-1β, which in turn provides positive feedback to induce synthesis of TNF-α by KCs8. In this study, 15d-PGJ2 reduced serum levels of TNF-α and IL-1β and reduced expression of F4/80, a major biomarker of KCs, as shown in Figure 2. It was hypothesized that the reduction of TNF-α and IL-1β in the 15d-PGJ2 treatment groups was due to the inhibitory effect on KC activation. Actually, 15d-PGJ2 has been proven to inhibit activation and recruitment of macrophages, both in vivo and in vitro34,35. Reduced activation of KCs, as well as reduced levels of pro-inflammatory cytokines, can result in a relatively mild inflammation with low serum ALT and AST levels and alleviated tissue damage, as shown in Figure 1.
Apoptosis can be mechanistically separated into intrinsic and extrinsic pathways depending on whether a trigger event is intrinsic or extrinsic to the cell. In hepatic I/R injury, the pro-inflammatory cytokine TNF-α, a major member of the death receptor ligands, activated the extrinsic apoptosis pathway as reported36. While mitochondrial damage caused by ROS resulted in a mitochondrial permeability transition, the release of cytochrome c into the cytosol led to an activation of the intrinsic apoptosis pathway7. In this study, the Bcl-2/Bax ratio was chosen to reflect mitochondrial apoptosis levels. Both Bcl-2 and Bax belong to the Bcl-2 family, which controls mitochondrial outer membrane permeabilization, cytochrome c release, and subsequent caspase activation37,38. Bcl-2 proteins are anti-apoptotic members of the Bcl-2 family that maintain mitochondrial membrane stability, while Bax is a pro-apoptotic member that disrupts mitochondrial membrane stability39. Although the cDNA level of both Bcl-2 and Bax increased in the I/R model group, the 15d-PGJ2 treatment groups showed a more elevated Bcl-2/Bax ratio, indicating a more stabilized mitochondrial membrane permeability. In addition, the Western blot results also revealed different expression at the protein level, as presented in Figure 3B. The reduced ROS expression in 15d-PGJ2 treatment groups compared with the I/R model group indicated a weak activation of intrinsic pathways. On the other hand, 15d-PGJ2 also helped to reduce TNF-α levels and thus weakened the extrinsic pathways. Therefore, it can be concluded that 15d-PGJ2 can reduce hepatic cell apoptosis by influencing both intrinsic and extrinsic pathways, as confirmed by the results of TUNEL staining (Figure 3C).
ROS refers to a class of generally short-lived, small, and highly oxygen-containing reactive molecules that include oxygen anions, free radicals, and hydrogen peroxide40. ROS are usually byproducts of mitochondrial respiration, and they can also be generated and transmitted by macrophages and neutrophils in inflammatory responses41. In addition to the pro-apoptotic effect caused by disruption of mitochondria function, ROS have recently been proven to induce autophagy in several ways, including via the HIF1α/BNIP3/Bcl-2 pathway3. HIF1α can be activated by ROS and induce transcription of BNIP3. The latter separates Bcl-2 from Beclin-1, thus activating Beclin-1, and initiating autophagy. Another biomarker of autophagy activation is the transformation from LC3-I to LC3-II, where the latter is only expressed on autophagosomes3. In addition, both expression of BNIP3 and LC3 can be induced by ROS through activating forkhead box O3 (FOXO3). Data in this study showed that cDNA levels of both BNIP3 and LC3 were reduced in the 15d-PGJ2 treatment groups (Figure 4A, 5A). In addition, the Western blot results also showed a reduction of LC3-II in the 15d-PGJ2 treatment groups, which indicated a decreased autophagy level (Figure 4B).
Studies have reported that, due to its special α,β-unsaturated carbonyl groups that act as an electrophilic center, 15d-PGJ2 could bind to the cysteine residue of Keap1, separate the Keap1-Nrf2 complex, and thus liberate Nrf2 from Keap1-dependent repression so that Nrf2 accumulates in the nucleus44. Indeed, our study observed and detected the nuclear expression of Nrf2 (Figure 5B, 5C). By transcribing a series of endogenous antioxidants, Nrf2 was regarded to be important in the clearance of ROS and prevention of oxidative stress. It can be speculated that the reduction of ROS may be related to the activation effect of 15d-PGJ2 on Nrf2. Kudoh et al demonstrated that 15d-PGJ2 showed no apparent reduction in the levels of aminotransferase and ROS in Nrf2 knock-out mice with I/R injury29, and the reduced ROS levels can be observed with fluorescence in the 15d-PGJ2 treatment groups when compared to I/R model groups, as shown in Figure 5C. As a result, the reduction of HIF1α in the nucleus can be observed by Western blot and immunohistochemistry in the 15d-PGJ2 treatment group, as shown in Figure 5B and 5C. Thus, it can be hypothesized that by inhibiting ROS generation and strengthening the clearance of ROS, 15d-PGJ2 can inhibit the HIF1α/BNIP3/Bcl-2 pathway and thus inhibit autophagy.
15d-PGJ2 is considered to be a natural ligand of PPARγ, which plays critical roles in the anti-inflammatory response45,46. Whether the protective effects of 15d-PGJ2 are associated with PPARγ needs to be considered. In our study, a significant increase in the serum levels of ALT and AST was observed, although no significant differences between the serum and protein expression of IL-1β and TNF-α were found with the GW9662 treatment. However, 15d-PGJ2 still showed a protective effect compared with the I/R group (P<0.05). In this regard, we consider the protective effects of 15d-PGJ2 on hepatic I/R injury to be partially PPARγ-dependent. This conclusion is similar to the result discovered by Kuboki et al, when they used PPARγ+/− mice47. The PPARγ+/− mice showed reduced activation of PPARγ and more severe liver injury after 8 h of reperfusion than their wild-type counterparts. However, the PPARγ pathway shows no effect on the reduction of pro-inflammatory cytokines by 15d-PGJ2 in our model (Figure 6B, 6C). Maier et al found that 15d-PGJ2 is capable of inhibiting the maturation of multiple inflammasomes, which is a vital step in IL-1β release, and this inhibition is independent of PPARγ, Nrf2, and cyclo-oxygenase 1 (COX-1)27. Our results also suggested that the reduction of TNF-α and IL-1β in the 15d-PGJ2 treatment groups was due to its inhibitory effect on the activation of KCs (Figure 2). The PPARγ agonists rosiglitazone and C-peptide were used, and their effects appeared to be limited to hepatocytes, as there was no effect on the production of TNF-α in previous studies47. These data indicate that PPARγ activation in hepatocytes has no effect on the reduction of pro-inflammatory cytokines by 15d-PGJ2. The specific protective mechanism of PPARγ on hepatic I/R injury needs to be further researched.
Overall, 15d-PGJ2 inhibits the activation of KCs in hepatic I/R injury, reducing the production of TNF-α and ROS, which causes hepatic cell necrosis and apoptosis. However, by activating Nrf2, 15d-PGJ2 also strengthens the clearance of ROS, consequently suppressing HIF1α/BNIP3 and LC3 expression and inhibiting autophagy. The probable mechanism by which 15d-PGJ2 reduced apoptosis and autophagy in mice of the I/R model is summarized in Figure 7.

Mechanism of 15d-PGJ2 protective effect. 15d-PGJ2 inhibits the activation of KCs in a hepatic I/R injury, reducing production of TNF-α and ROS which cause hepatic cell necrosis and apoptosis. On the other hand, by activating Nrf2, 15d-PGJ2 also strengthens clearance of ROS, consequently suppresses HIF1β/BNIP3/Bcl-2 and inhibits autophagy.
Conclusion
In this study, we confirmed the protective effect of 15d-PGJ2 on a model of hepatic I/R injury in mice. The effect may rely on a reduction in the activation of KCs and on activation of the Nrf2 pathway, which lead to inhibition of ROS generation, apoptosis, and autophagy.
Author contribution
Kan CHEN, Ying-qun ZHOU and Chuan-yong GUO designed research; Kan CHEN, Jing-jing LI, Sai-nan LI, Jiao FENG, Tong LIU and Fan WANG performed research; Wei-qi DAI, Yu-jing XIA and Jie LU contributed new reagents or analytic tools; Jing-jing LI and Sai-nan LI analyzed data; Kan CHEN wrote the paper.

(D) ROS level was detected by ROS Fluorescent Probe-DHE, and the results, calculated with IOD value, clearly showed the difference among all groups. n=6, *P<0.05 for NC vs I/R. #P<0.05 for I/R vs I/R+15d-PGJ2.
References
Kim HY, Kim SJ, Lee SM . Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J 2015; 282: 259–70.
Shen M, Lu J, Dai W, Wang F, Xu L, Chen K, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm 2013; 2013: 461536.
Cursio R, Colosetti P, Gugenheim J . Autophagy and liver ischemia-reperfusion injury. Biomed Res Int 2015; 2015: 417590.
Wang C, Chen K, Xia Y, Dai W, Wang F, Shen M, et al. N-Acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One 2014; 9: e108855.
Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA . Post-injury multiple organ failure: the role of the gut. Shock 2001; 15: 1–10.
Belghiti J, Hiramatsu K, Benoist S, Massault P, Sauvanet A, Farges O . Seven hundred forty-seven hepatectomies in the 1990s: an update to evaluate the actual risk of liver resection. J Am Coll Surg 2000; 191: 38–46.
Dogan S, Aslan M . Hepatic ischemia-reperfusion injury and therapeutic strategies to alleviate cellular damage. Hepatol Res 2011; 41: 103–17.
Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, et al. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock 1996; 5: 34–40.
Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA . The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology 1996; 23: 506–14.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–91.
Li J, Wang F, Xia Y, Dai W, Chen K, Li S, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs 2015; 13: 3368–87.
D'Autreaux B, Toledano MB . ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007; 8: 813–24.
Essani NA, McGuire GM, Manning AM, Jaeschke H . Endotoxin-induced activation of the nuclear transcription factor kappa B and expression of E-selectin messenger RNA in hepatocytes, Kupffer cells, and endothelial cells in vivo. J Immunol 1996; 156: 2956–63.
Walker NI, Harmon BV, Gobe GC, Kerr JF . Patterns of cell death. Methods Achiev Exp Pathol 1988; 13: 18–54.
Festjens N, Vanden BT, Vandenabeele P . Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 2006; 1757: 1371–87.
Cheng P, Wang F, Chen K, Shen M, Dai W, Xu L, et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediators Inflamm 2014; 2014: 935251.
Nishida K, Yamaguchi O, Otsu K . Crosstalk between autophagy and apoptosis in heart disease. Circ Res 2008; 103: 343–51.
Cohen GM, Sun XM, Fearnhead H, MacFarlane M, Brown DG, Snowden RT, et al. Formation of large molecular weight fragments of DNA is a key committed step of apoptosis in thymocytes. J Immunol 1994; 153: 507–16.
Murrow L, Debnath J . Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 2013; 8: 105–37.
Shen M, Chen K, Lu J, Cheng P, Xu L, Dai W, et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-beta1 expression and autophagy. Mediators Inflamm 2014; 2014: 954502.
Li L, Tan J, Miao Y, Lei P, Zhang Q . ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 2015; 35: 615–21.
Wang C, Xia Y, Zheng Y, Dai W, Wang F, Chen K, et al. Protective effects of N-acetylcysteine in concanavalin A-induced hepatitis in mice. Mediators Inflamm 2015; 2015: 189785.
Li S, Xia Y, Chen K, Li J, Liu T, Wang F, et al. Epigallocatechin-3-gallate attenuates apoptosis and autophagy in concanavalin A-induced hepatitis by inhibiting BNIP3. Drug Des Devel Ther 2016; 10: 631–47.
Kaspar JW, Niture SK, Jaiswal AK . Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med 2009; 47: 1304–9.
Tsuchihashi S, Fondevila C, Kupiec-Weglinski JW . Heme oxygenase system in ischemia and reperfusion injury. Ann Transplant 2004; 9: 84–7.
Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A 2004; 101: 2040–5.
Maier NK, Leppla SH, Moayeri M . The cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomes. J Immunol 2015; 194: 2776–85.
Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, et al. 15-Deoxy-Delta12,14-prostaglandin J2, an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem Pharmacol 2011; 82: 1335–51.
Kudoh K, Uchinami H, Yoshioka M, Seki E, Yamamoto Y . Nrf2 activation protects the liver from ischemia/reperfusion injury in mice. Ann Surg 2014; 260: 118–27.
Kang ES, Hwang JS, Ham SA, Park MH, Kim GH, Paek KS . 15-Deoxy-Delta12,14-prostaglandin J2 prevents oxidative injury by upregulating the expression of aldose reductase in vascular smooth muscle cells. Free Radic Res 2014; 48: 218–29.
Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA . Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res 2008; 147: 153–9.
Carden DL, Granger DN . Pathophysiology of ischaemia-reperfusion injury. J Pathol 2000; 190: 255–66.
Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ . Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun 1991; 15: 277–84.
Liu X, Yu H, Yang L, Li C, Li L . 15-Deoxy-Delta12,14-prostaglandin J2 attenuates the biological activities of monocyte/macrophage cell lines. Eur J Cell Biol 2012; 91: 654–61.
Han Z, Zhu T, Liu X, Li C, Yue S, Liu X, et al. 15-deoxy-Delta12,14-prostaglandin J2 reduces recruitment of bone marrow-derived monocyte/macrophages in chronic liver injury in mice. Hepatology 2012; 56: 350–60.
Li J, Xia Y, Liu T, Wang J, Dai W, Wang F, et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS One 2015; 10: e120440.
Luedde T, Kaplowitz N, Schwabe RF . Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014; 147: 765–83.
Shen M, Lu J, Cheng P, Lin C, Dai W, Wang F, et al. Ethyl pyruvate pretreatment attenuates concanavalin a-induced autoimmune hepatitis in mice. PLoS One 2014; 9: e87977.
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012; 45: 487–98.
Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S . Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int 2014; 2014: 761264.
Chakrabarti S, Jahandideh F, Wu J . Food-derived bioactive peptides on inflammation and oxidative stress. Biomed Res Int 2014; 2014: 608979.
Cheng P, Chen K, Xia Y, Dai W, Wang F, Shen M, et al. Hydrogen sulfide, a potential novel drug, attenuates concanavalin A-induced hepatitis. Drug Des Devel Ther 2014; 8: 1277–86.
Mahalingaiah PK, Singh KP . Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS One 2014; 9: e87371.
Haskew-Layton RE, Payappilly JB, Xu H, Bennett SA, Ratan RR . 15-Deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARgamma. J Neurochem 2013; 124: 536–47.
Li C, Yang H, Hou Y, Chiu Y, Chiu W . Dietary fish oil reduces systemic inflammation and ameliorates sepsis-induced liver injury by up-regulating the peroxisome proliferator-activated receptor gamma-mediated pathway in septic mice. J Nutr Biochem 2014; 25: 19–25.
Xu F, Li J, Ni W, Shen Y, Zhang X . Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS One 2013; 8: e55080.
Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, et al. Peroxisome proliferator-activated receptor-γ protects against hepatic ischemia/reperfusion injury in mice. Hepatology 2008; 47: 215–24.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81270515 and No 81500466).
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
About this article

Cite this article
Chen, K., Li, Jj., Li, Sn. et al. 15-Deoxy-Δ12,14-prostaglandin J2 alleviates hepatic ischemia-reperfusion injury in mice via inducing antioxidant response and inhibiting apoptosis and autophagy. Acta Pharmacol Sin 38, 672–687 (2017). https://doi.org/10.1038/aps.2016.108
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.108
Keywords
This article is cited by
-
Autophagy in hepatic ischemia–reperfusion injury
Cell Death Discovery (2023)
-
The liver protection of propylene glycol alginate sodium sulfate preconditioning against ischemia reperfusion injury: focusing MAPK pathway activity
Scientific Reports (2017)
