
Abstract
Aim:
Compound 10b is a hybrid molecule of edaravone and a ring-opening derivative of 3-n-butylphthalide (NBP). The aim of this study was to examine the effects of compound 10b on brain damage in rats after focal cerebral ischemia.
Methods:
SD rats were subjected to 2-h-middle cerebral artery occlusion (MCAO). At the onset of reperfusion, the rats were orally treated with NBP (60 mg/kg), edaravone (3 mg/kg), NBP (60 mg/kg)+edaravone (3 mg/kg), or compound 10b (70, 140 mg/kg). The infarct volume, motor behavior deficits, brain water content, histopathological alterations, and activity of GSH, SOD, and MDA were analyzed 24 h after reperfusion. The levels of relevant proteins in the ipsilateral striatum were examined using immunoblotting.
Results:
Administration of compound 10b (70 or 140 mg/kg) significantly reduced the infarct volume and neurological deficits in MCAO rats. The neuroprotective effects of compound 10b were more pronounced compared to NBP, edaravone or NBP+edaravone. Furthermore, compound 10b significantly upregulated the protein levels of the cytoprotective molecules Bcl-2, HO-1, Nrf2, Trx, P-NF-κB p65, and IκB-α, while decreasing the expression of Bax, caspase 3, caspase 9, Txnip, NF-κB p65, and P-IκB-α.
Conclusion:
Oral administration of compound 10b effectively attenuates rat cerebral ischemia injury.
Similar content being viewed by others


Introduction
Cerebral ischemia is a leading cause of morbidity and one of the most common causes of death worldwide. This condition is associated with cardiovascular disease and cancer, and its incidence increases with age1. Mechanisms implicated in the pathophysiology of ischemia–reperfusion (I/R) injury include excitotoxicity, oxidative stress, intracellular calcium overload and cell death2,3,4.
Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, was approved by the Japanese Ministry of Health in 2001 for the treatment of ischemic stroke. Edaravone exerts neuroprotective effects by scavenging free radicals, inhibiting lipid peroxidation and oxidative damage to brain cells, endothelial cells and nerve cells, and reducing the effects of cerebral ischemia and edema, thus decreasing the tissue damage caused by acute cerebral infarction5,6,7,8. Given that the anionic form of edaravone may also scavenge free radicals using the one-electron transfer mechanism involved in protecting nerve cells, we hypothesize that derivatives of edaravone might be potential agents for therapeutic intervention following ischemic stroke.
Compound 10b, a novel derivative of edaravone, is a hybrid of edaravone and a ring-opening derivative of 3-n-butylphthalide (NBP) (Figure 1). Racemic NBP, a natural compound extracted from celery seed, was approved for the treatment of ischemic stroke by the State Food and Drug Administration (SFDA) of China in 2002. It has been reported that NBP has a variety of biological activities, including suppressing platelet aggregation and thrombosis, improving cerebral microcirculation and reducing infarct volume, all of which are associated with ischemic stroke9,10,11,12. We recently reported in a previous study that compound 10b displayed enhanced antiplatelet aggregation effects and protection against nerve cell damage via the synergistic action of NBP and edaravone in vitro13. Increasing focus has therefore been put on the therapeutic potential of compound 10b for cerebral ischemia injury.

Chemical structures of 10b, 3-n-butylphthalide (NBP) and 3-methyl-1-phenyl-2-pyrazolin-5-one (edaravone).
Therefore, the aims of the present study were to investigate whether compound 10b can provide improved protective effects on ischemic neuronal injury in vivo and to explore the possible mitochondrial mechanisms that may underlie the neuroprotective effects.
Materials and methods
Materials
Compound 10b and NBP were kindly provided by the Center of Drug Discovery at China Pharmaceutical University. The purity of 10b and NBP was more than 95%. These compounds were dissolved in 0.5% sodium carboxyl methyl cellulose (CMC-Na).
Animal experiments and drug treatment
We investigated the anti-ischemic activity of compound 10b in a rat model of transient focal cerebral ischemia by intraluminal occlusion of the middle cerebral artery (MCAO) for 2 h followed by recirculation, which has been widely used to evaluate the protective effects of antistroke agents14. Adult male Sprague-Dawley rats (57–61 d old and weighing 250–280 g) were purchased from B&K Universal Group Limited (Shanghai, China). The rats were housed at 22–24 °C with light from 8:00 am to 8:00 pm. All animals were fed a commercial diet and had free access to water during the experiment.
The rats were anesthetized with chloral hydrate (300 mg/kg, ip) and subjected to MCAO as described previously15, with slight modifications. The right common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were briefly exposed. A 4–0 monofilament nylon suture (Beijing Sunbio Biotech Co, Ltd, Beijing, China) with a rounded tip was inserted into the ICA through the ECA stump and gently advanced to occlude the MCA. After 2 h of MCAO, the suture was withdrawn to restore blood flow (reperfusion). The same surgery was performed, omitting the MCA occlusion, in sham-operated rats. The core body temperatures were monitored with a rectal probe and were maintained at 37±0.5 °C during the entire procedure. The rats were randomly assigned to seven groups (12 rats per group), consisting of the sham group (0.5% CMC-Na, ig), the MCAO group (0.5% CMC-Na, ig), the NBP group (60 mg/kg NBP, ig), the edaravone group (3.0 mg/kg edaravone, ig), the NBP+edaravone group (60 mg/kg NBP+3.0 mg/kg edaravone, ig), the low-dose compound 10b group (70 mg/kg, the dose of 10b being equivalent to that of NBP+Eda, ig), and the high-dose compound 10b group (140 mg/kg, the dose of 10b was equimolar to that of NBP+Eda, ig). The drugs were administered at the onset of reperfusion which lasted for 24 h.
Neurological scoring
The neurological function was scored by a blinded observer 24 h after reperfusion. Briefly, the neurological deficits were graded according to the following scale: 0, no deficit; 1, forelimb weakness; 2, circling to affected side; 3, inability to bear weight on the affected side; and 4, no spontaneous motor activity16.
Measurement of infarct size
At 24 h after reperfusion, the rats (n=6) were decapitated, and the brains were rapidly removed, placed on ice, and sliced into six 2-mm-thick coronal sections. The sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (Sigma Chemical Co, St Louis, MO, USA) for 30 min at 37 °C followed by overnight immersion in 10% formalin. The infarcted tissue remained unstained (white), whereas normal tissue was stained red. The infarct zone was demarcated and analyzed using Image-Pro Plus 6 (Media Cybernetics, Bethesda, MD, USA). The infarcted area was expressed as a percentage of the total area of the brain.
Measurement of brain water content
At 24 h after reperfusion, some rats from each group (n=6) were sacrificed, and the brains were rapidly removed onto ice. Ischemic hemispheres were then isolated from the brains. The tissue samples were immediately weighed to obtain the wet weight. The brains were then dried at 100 °C for 24 h to determine their dry weight. The percentage of brain water content was calculated as (wet weight-dry weight)/wet weight×100%.
Histopathological examination
At 24 h after reperfusion, the rats were deeply anesthetized with chloral hydrate and perfused with heparinized PBS, followed by perfusion with 4% paraformaldehyde in PBS. The brain tissue samples were cut coronally into three blocks from the optic chiasm to the infundibulum of the hypophysis. The middle blocks were embedded in paraffin. The paraffin-embedded tissues were sectioned at 5 μm, and the brain tissue sections were stained with hematoxylin and eosin (HE). Histopathological evaluation was performed by pathologists in a blinded manner17.
Glutathione (GSH) activity assay
Twenty-four hours after MCAO, the rats were sacrificed and the brains were removed. The GSH activity in homogenates extracted from the cortex of the ischemic hemisphere of the brain were measured. The antioxidant enzyme activity of GSH was determined using a modified version of a previously described method18. The reaction mixture was prepared by combining 600 μL of potassium phosphate buffer (0.05 mol/L, pH 7.0, 0.1 mmol/L EDTA), 100 μL of 10 mmol/L GSH (reduced form), 100 μL of 1.5 mmol/L NADPH, and 100 μL of the brain homogenate supernatant. A 100 μL aliquot of brain homogenate supernatant was added to the reaction mixture and incubated at 37 °C for 10 min. Fifty microliter of 12 mmol/L t-butyl hydroperoxide was added to this mixture. The change in absorbance was measured at 340 nm for 1 min. One unit of activity is equal to 1 mmol of NADPH oxidized/mg protein/min. The GSH activity was expressed as a fold change relative to the sham group.
Superoxide dismutase (SOD) activity assay
Twenty-four hours after MCAO, the rats were sacrificed and the brains were removed. The SOD activity in homogenates extracted from the cortex of the ischemic hemisphere of the brain were measured. The SOD activity was measured based on the extent of inhibition of amino blue tetrazolium formazan formation in the mixture containing nicotinamide adenine dinucleotide, phenazine methosulphate, and nitroblue tetrazolium (NADH-PMSNBT), as previously described19,20. The assay mixture contained 0.1 mL of the brain homogenate supernatant, 1.2 mL of sodium pyrophosphate buffer (pH 8.3, 0.052 mol/L), 0.1 mL of PMS (186 μmol/L), 0.3 mL of NBT (300 μmol/L), and 0.2 mL of NADH (750 μmol/L). The reaction was started by the addition of NADH. After incubation at 30 °C for 90 s, the reaction was stopped by the addition of 0.1 mL of glacial acetic acid. The reaction mixture was stirred vigorously with 4.0 mL of n-butanol. The intensity of chromogen color in the butanol was measured spectrophotometrically at 560 nm. The SOD activity was calculated according to the SOD standard control and expressed as a fold change relative to the sham group.
Lipid peroxidation level measurement
Twenty-four hours after MCAO, the rats were sacrificed and the brains were removed. The MDA activity in homogenates extracted from the cortex of the ischemic hemisphere of the brain was measured. Lipid peroxidation, a major indicator of oxidative stress, was determined by measuring the malondialdehyde (MDA) level in the tissue homogenates. MDA is an end product of lipid peroxidation, and its level was determined spectrophotometrically using the thiobarbituric acid-reactive substances method, with little modification21,22. Briefly, 5 μL supernatant was mixed with 25 μL 3.0% sodium dodecylsulfate, 75 μL acetic acid buffer (pH 3.5), 75 μL 0.8% 2-thiobarbituric acid, and 20 μL deionized water followed by heating in a water bath at 95 °C for 1 h. After cooling, 250 μL of a mixture of n-butanol and pyridine (15:1) was added, and the mixture was centrifuged at 1400×g for 15 min. The organic phase was collected, and the fluorescence intensity was measured with excitation at 515 nm and emission at 553 nm. A standard curve was run simultaneously with each set of samples by using 1,1,3,3-tetramethoxypropane as an external standard. The MDA content was expressed as a fold change relative to the sham group.
Western blot analysis
Extraction of protein in the cytosolic, mitochondrial and nuclear fractions was isolated by multiple centrifugation using a mitochondria/nuclei isolation kit (KGA828, Nanjing KeyGEN Biotechnology Co, Ltd, Nanjing, China). The protein concentrations were determined using a bicinchoninic acid assay. Equal amounts of protein (40 μg/lane) were subjected to SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore Corporation, Boston, MA, USA). The blots were blocked for 2 h in 5% nonfat dry milk-TBS-0.1% Tween 20 and then washed. The blots were incubated overnight with primary antibodies at 4 °C followed by a 2-h incubation with a horseradish peroxidase-conjugated secondary anti-rabbit antibody (1:10 000; Bioworld Technology Co, Ltd, Saint Louis, MN, USA). A gel imaging system (ChemiScope 2850, Clinx Science Instruments Co, Ltd, Shanghai, China) was used to detect immunoreactivity as indicated by enhanced chemiluminescence detection reagents (KGP1123, Nanjing Key-GEN Biotechnology Co, Ltd, Nanjing, China). The primary antibodies used were polyclonal antibodies against Txnip (ab86983) (1:500; Abcam Co, Ltd, Cambridge, Cambs, UK), Trx (C63C6), P-NF-κB p65 (Ser536), NF-κB p65 (C22B4), P-IκB-α, IκB-α, GAPDH (Cell Signaling Technology Co, Ltd, Beverly, MA, USA), Bcl-2, Bax, caspase 3 (Asp175), caspase 9 (Asp353) COX IV (3E11) (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), HO-1 (P249), Nrf2 (D1C9) (1:500; Bioworld Technology Co, Ltd, Saint Louis, MN, USA), and β-actin (1:5000; Bioworld Technology Co, Ltd, Saint Louis, MN, USA).
Statistical analysis
The data are expressed as the mean±SD. Statistical analyses were performed using one-way ANOVA followed by Tukey's multiple comparison test for multiple comparisons. A P value of less than 0.05 was considered to be statistically significant.
Results
Compound 10b attenuates the neurological deficits after I/R in Rats
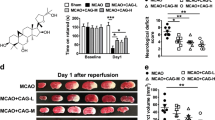
The neurological function of the individual rats was evaluated using Longa's method 24 h after reperfusion. As shown in Figure 2A, no obvious neurological deficit was observed in the sham-operated, CMC-Na-treated animals. Compared with the rats in the sham-operated group, those in the I/R group showed prominent neurological deficits 24 h after reperfusion. Post-ischemic treatment with 10b (70 or 140 mg/kg) significantly improved neurobehavioral function, as indicated by the 32.26% and 38.71% reduction in neurological deficit scores, respectively, compared to the I/R group. The neuroprotective effect of the high dose of compound 10b was more potent than that of NBP (22.58%), Eda (29.03%), or NBP+Eda (32.26%).

Effect of compound 10b on brain injury of cerebral I/R rats. (A) Neurological deficits were assessed at 24 h after reperfusion (n=12). (B) The infarcted brain areas were visualized using TTC staining. A representative example is shown for each treatment group. (C) Quantitative analysis of the infracted brain regions. The ratio of the infarcted area to the whole brain area in individual rat was calculated (n=6). (D) Effects of 10b on the brain water content after I/R (n=6). The data are expressed as the mean±SD. cP<0.01 compared with the sham group. eP<0.05, fP<0.01 compared with the I/R group.
Reduction of infarct size and brain water content in ischemic brains
The brain infarct size in the individual rats was evaluated by the 2,3,5-triphenyltetrazolium chloride (TTC) assay. There was no infarct damage in the sham group rats (Figure 2B). In comparison with the untreated I/R group, the infarct size in the 10b-treated rats was reduced by 52.80% (10b at 70 mg/kg) and 61.77% (10b at 140 mg/kg) (Figure 2C). Furthermore, the effects of 10b at 70 mg/kg or 140 mg/kg were more pronounced than those of NBP (34.41%), Eda (38.47%), and NBP+Eda (46.45%). Additionally, treatment with low or high doses of 10b, NBP, Eda, or NBP+Eda markedly reduced the brain water content of ischemic rats (Figure 2D).
Attenuation of cerebral damage
Histopathological analysis revealed that treatment with 10b considerably attenuated the cerebral damage in rats compared with that of I/R-treated model rats. As shown in Figure 3, HE staining of the ipsilateral hemisphere of ischemic brains showed cellular edema, neuronal perikarya shrinkage, and macrophage infiltration, whereas cellular edema in the drug-treated rats was not obvious, especially in the animals treated with compound 10b at 140 mg/kg.

The cortex of injured cerebral hemisphere was stained with HE and examined under a light microscope after I/R.
Compound 10b upregulates endogenous antioxidant systems following cerebral ischemia/reperfusion
To further explore the mechanism underlying the anti-ischemic effect of 10b, we evaluated the levels of glutathione (GSH) and malondialdehyde (MDA) and the activity of superoxide dismutase (SOD) in the brain tissues. Compared with the sham group rats, the levels of GSH in the I/R group were decreased by 52.56% (from 167.22 to 79.33 nmol/mg). However, post-ischemic treatment with 10b (70 or 140 mg/kg) significantly enhanced the GSH levels by approximately 80.64% or 97.30% to 143.30 or 156.52 nmol/mg, respectively, which were slightly higher than those in the NBP+Eda-treated rats (69.60%, 134.54 nmol/mg) and significantly higher than those in the NBP-treated rats (57.15%, 124.67 nmol/mg) or the Eda-treated rats (61.09%, 127.80 nmol/mg ) (Figure 4A).

Effect of 10b on the level of brain GSH (A), SOD activity (B) and MDA (C) in the ischemic cerebral cortex of rats after I/R. The data are expressed as the mean±SD of the individual groups of rats (n=6) and were analyzed by one-way analysis of variance (ANOVA) followed by a post-hoc Tukey test. cP<0.01 compared with the sham group. fP<0.01 compared with the I/R group.
Furthermore, the SOD activity in the ischemic brains from the I/R group of rats was significantly decreased by 43.15% (from 182.65 to 103.84 U/mg, Figure 4B) relative to that in the sham-operated rats. By contrast, treatment with compound 10b at 70 or 140 mg/kg significantly increased the SOD activity by 55.44% (from 103.84 to 161.41 U/mg) and 73.70% (from 103.84 to 180.37 U/mg), respectively, compared with that in the I/R group. Notably, the SOD activity in rats treated with 10b at 70 or 140 mg/kg was slightly greater than that of NBP+Eda-treated (46.70%, 152.33 U/mg) group and significantly greater than that in the NBP-treated rats (35.40%, 140.60 U/mg) or the Eda-treated rats (38.45%, 143.76 U/mg).
Further analysis revealed that the MDA content of the brain, an indicator of lipid peroxidation, was significantly elevated by 124.26% (from 8.45 to 18.95 nmol/mg) in the I/R group compared with that in the sham-operated animals. However, post-ischemic treatment with 10b at 70 or 140 mg/kg remarkably decreased the levels of brain MDA by 33.83% (from 18.95 to 12.54 nmol/mg) and 57.10% (from 18.95 to 8.13 nmol/mg), respectively, compared with that in the I/R group (Figure 4C). More importantly, the effect of high-dose 10b on the brain lipid peroxidation was much stronger than that of NBP (22.59%, 14.67 nmol/mg), Eda (25.35%, 14.15 nmol/mg) or NBP+Eda (33.98%, 12.51 nmol/mg).
Nuclear factor-E2-related factor (Nrf2) is a key transcription factor that regulates antioxidant genes as an adaptive response to oxidative stress or pharmacological stimuli23,24. To investigate the effect of compound 10b treatment on the Nrf2 signaling, the cellular locations of Nrf2 were evaluated after I/R. Western blot analysis revealed that the expression of Nrf2 in the nuclear fraction was significantly higher in the 10b (70 or 140 mg/kg), NBP, Eda and NBP+Eda groups than that in the I/R group (Figure 5B). These results demonstrated that 10b treatment induced Nrf2 nuclear localization.

Compound 10b upregulates endogenous antioxidant systems following cerebral I/R. (A) The expression of Nrf2 in the cortex was examined by Western blot. β-Actin was used as the loading control. (B) Statistical results from the densitometric measurements after normalization to β-actin were represented as the mean±SD (n=6 for each group). (C) The representative immunoblots of HO-1 in cerebral cortex. β-Actin was used as the loading control. (D) Statistical results from the densitometric measurements after normalization to β-actin were represented as the mean±SD (n=6 for each group). (E) Representative immunoblots of Trx and Txnip. GAPDH was used as the loading control. (F) Statistical results from the densitometric measurements after normalization to GAPDH were represented as the mean±SD (n=6 for each group). Significance: bP<0.05, cP<0.01 compared with the sham group; eP<0.05, fP<0.01 compared with the I/R group by one-way ANOVA followed by Tukey's multiple comparison test.
Moreover, HO-1 has a key role in the resistance of the neuronal cells to ROS-induced cell death. To evaluate the effects of 10b treatment on antioxidant enzymes, the cellular locations of HO-1 were measured after I/R. Compound 10b treatment (70 or 140 mg/kg) significantly increased the expression of HO-1 (Figure 5D). An increase in HO-1 expression was also observed in the NBP+Eda-treated group.
Protection from ROS is mediated by superoxide dismutase and the glutathione and thioredoxin (Trx) systems25,26. We therefore measured the expression of Trx and Txnip by Western blot analysis. The changes in Trx and Txnip expression after I/R were analyzed in the I/R groups using immunoblotting. Compared with the I/R group, significant increases in Trx expression were observed in the 10b-treated (70 or 140 mg/kg), NBP-treated, Eda-treated and NBP+Eda-treated group after I/R (Figure 5Fa). The Txnip level appeared to decrease slightly in the NBP-treated group and more prominently in the 10b-treated (70 or 140 mg/kg) groups, the Eda-treated group and the NBP+Eda-treated group (Figure 5Fb).
Overall, these observations indicate that compound 10b treatment activates Nrf2, HO-1, and Trx, restrains Txnip, and upregulates antioxidant enzymes against I/R-induced neuronal injury.
Compound 10b inhibits apoptosis following cerebral ischemia/reperfusion
The proto-oncoproteins (Bcl-2 and Bax) are key regulators of the mitochondrial apoptotic pathway initiated by a variety of extracellular and intracellular stressors27,28,29. As shown in Figure 6B, NBP treatment, Eda treatment, NBP+Eda treatment and 10b (70 or 140 mg/kg) treatment significantly increased the expression of Bcl-2 in mitochondria and inhibited the translocation of Bax to the mitochondria. Caspases are widely expressed in inactive proenzyme forms in normal cells. Active/cleaved caspases often activate other pro-caspases, allowing initiation of a protease cascade in apoptotic cells30. Caspase 9 and caspase 3, markers of the mitochondrial apoptotic pathways, were selected to evaluate the effect of 10b treatment on the caspase cascade. As shown in Figure 6D, caspase 9 and caspase 3 were activated in the I/R group. However, both 10b (70 or 140 mg/kg) and the positive drugs (60 mg/kg NBP, 3 mg/kg Eda, and 60 mg/kg NBP+3 mg/kg Eda) effectively suppressed the I/R-induced caspase activation, which was decreased compared with the I/R group.

Effects of compound 10b on the mitochondrial apoptotic pathway in the cerebral cortex after I/R. (A) Representative immunoblots of Bcl-2 and Bax in mitochondria. COX IV was used as the loading control. (B) Quantitation of the Western blot analysis for Bcl-2 and Bax normalized to COX IV. The data are expressed as the mean±SD (n=6 for each group). (C) Representative immunoblots of caspase 9 and caspase 3. β-Actin was used as the loading control. (D) Statistical results from the densitometric measurements after normalization to β-actin were calculated as the mean±SD (n=6 for each group). Significance: cP<0.01 compared with the sham group; eP<0.05, fP<0.01 compared with the I/R group by one-way ANOVA followed by Tukey's multiple comparison test.
Effects of 10b treatment on protein expression of NF-κB and IκB-α following cerebral ischemia/reperfusion
Previous studies have reported that NF-κB is associated with cell proliferation and apoptosis, and expression of IκB-α is well known to block NF-κB activation by conferring resistance to TNF-induced degradation31,32,33. We next examined whether NF-κB and IκB-α participated in the neuroprotective effect of 10b in vitro. Consequently, Western blot analysis of the I/R group showed an increase in the expression of NF-κB p65, in contrast to the change in expression of IκB-α. However, 10b (70 or 140 mg/kg), NBP, Eda and NBP+Eda treatment mitigated the increased NF-κB p65 and augmented the reduced IκB-α (Figure 7Ba, 7Bc). Moreover, NBP+Eda treatment also increased the expression of IκB-α. The expression of P-NF-κB was significantly decreased in the I/R group, while 10b (140 mg/kg) treatment markedly increased the expression of P-NF-κB, whereas the expression of P-NF-κB was not influenced by NBP treatment, Eda treatment or NBP+Eda treatment (Figure 7Bb). In addition, the expression of P-IκB-α was upregulated in the I/R group, and 10b (70 or 140 mg/kg) treatment augmented it markedly (Figure 7Bd). These results indicate that 10b treatment regulates the expression of NF-κB p65 and rescues the expression of IκB-α to attenuate the neuronal injury following I/R. These results indicate the involvement of activation of NF-κB and TNF-α signaling pathways in the neuroprotective effect of 10b.

Compound 10b regulates the NF-κB/TNF-α signaling pathway. (A) Representative immunoblots of total and phosphorylated NF-κB p65 and IκB-α in the cortex of ischemic hemisphere measured after I/R using Western blot. GAPDH was used as the loading control. (B) Statistical results from the densitometric measurements after normalization to GAPDH were represented as the mean±SD (n=6 for each group). cP<0.01 compared with sham group; eP<0.05, fP<0.01 compared with I/R group by one-way ANOVA followed by Tukey's multiple comparison test.
Discussion
In the current study, our data demonstrated that compound 10b protected the brain against I/R injury in a rat MCAO model. We found that treatment with 10b at 70 and 140 mg/kg can provide substantial synergistic neuroprotection on focal cerebral ischemia compared with NBP treatment (60 mg/kg), edaravone treatment (3 mg/kg) or co-administration of NBP (60 mg/kg) and edaravone (3 mg/kg). These results show that the MCAO rats had significant motor behavioral deficits and extensive infarction in the ipsilateral cerebral cortical and subcortical areas throughout a series of brain sections. In the groups treated with NBP, Eda, or NBP+Eda, a decrease in the infarct volume and brain water content and an improvement in the neurological deficits were observed, but greater effects were seen in the rats that were given 10b (140 mg/kg) (Figure 2). HE staining also confirmed that 10b treatment, NBP treatment, Eda treatment and NBP+Eda treatment rescued neuronal loss after ischemia. In addition, the degree of neuroprotection induced by 10b (70 or 140 mg/kg) treatment was greater than that of NBP treatment, Eda treatment and NBP+Eda treatment in the same I/R model (Figure 3). These results suggest that 10b might mimic the synergistic combination therapy of NBP and edaravone reported in the previous study13. Thus, we concluded that 10b treatment may induce a stronger degree of neuroprotection and prevention of cellular edema during focal cerebral ischemia compared with equivalent NBP+Eda treatment. At a dose equimolar to the NBP+Eda treatment, 10b also produced greater effects of reduction in infarct volume, brain water content and improvement of neurological deficits.
Most of these drugs exert protective effects via their ability to upregulate the cellular antioxidant defense systems. The regulation of most of these antioxidant enzymes occurs through activation of Nrf234. Previous studies have shown that Nrf2 translocates from the cytosol to the nucleus and binds to the antioxidant responsive element (ARE)35, a cis-acting regulatory element or enhancer sequence, found in the promoter region of certain genes, including glutathione synthase, HO-1, and catalase36. The coordinated action of Nrf2 mediates the response to oxidative stress37. Compound 10b treatment mitigated oxidative stress as evidenced by the decreased levels of MDA (Figure 4C). This was the first study to report that 10b treatment significantly induced the translocation of Nrf2 and HO-1 to the nucleus (Figure 5B and 5D) and markedly increased the cellular antioxidant defense systems including those of GSH, SOD, and HO-1 (Figure 4A, 4B and 5D). The data confirm that Nrf2 plays a fundamental role in neuroprotection induced by 10b treatment (Figure 5B). The major finding of the current study supports the idea that 10b treatment can induce a signaling mechanism to combat oxidative stress, as evidenced by the ability of 10b treatment to increase cellular antioxidants and substantially reduce intracellular ROS production via the Nrf2 signaling pathway. Specifically, compared with treatment with NBP+Eda, treatment with equimolar 10b can significantly induce the suppression of MDA, translocation of Nrf2 and HO-1 to the nucleus and markedly increase the cellular antioxidant defense systems including those of GSH, SOD, and HO-1.
In addition, we determined whether the levels of the endogenous proteins Trx and Txnip are modulated in response to 10b treatment. Immunoblot analysis showed a significant decrease in Txnip expression in the 10b-treated (70 or 140 mg/kg) rats compared with those in the I/R group (Figure 5Fb). The Trx level, on the other hand, was markedly increased in the NBP-treated, Eda-treated and NBP+Eda-treated rats. Trx upregulation was more evident in the 10b-treated group (Figure 5Fa). Considerable 10b-induced changes were observed in Txnip protein levels. Txnip is involved in redox regulation through interaction with the catalytic active center of Trx and downregulates its activity and expression38,39. Thus, Txnip is considered to be a negative regulator of Trx and redox. Modulation of the Txnip and Trx levels after I/R observed in this study agree with the published data39. The compound 10b-induced reduction of Txnip expression is most likely responsible for the reduced upregulation of Trx. These changes in Txnip and Trx levels, which are predominantly affected by 10b treatment, are particularly interesting. These results show that in the group treated with 10b at a dose of equal molarity to NBP+Eda-treatment, there was a significant decrease in the Txnip expression and a marked increase in the Trx protein levels in contrast with the effects of the NBP+Eda treatment.
Regarding mitochondrial protection, 10b sequentially inhibited both caspase-dependent and caspase-independent mitochondrial cell death pathways. By suppressing both Bax translocation to the mitochondrial membrane and the downregulation of Bcl-2 induced by I/R, 10b could block subsequent caspase activation, thus partially preventing apoptotic cell death. Compared with the NBP group or the Eda group (Figure 6B), the increase of Bcl-2 level is more prominent in the 10b group (Figure 6Ba). Moreover, based on the recent observation that Bcl-2 regulates both Ca2+ release from the endoplasmic reticulum40 and the essential pool of mitochondrial GSH41, we propose that 10b could maintain [Ca2+]i homeostasis and enhance the mitochondrial antioxidant status by upregulating Bcl-2. In contrast to 10b, the other treatments (NBP, Eda and NBP+Eda) had no significant effect on the Bax level (Figure 6Bb). Thus, it seems reasonable to speculate that 10b blocks Bax translocation. It follows that 10b could more strongly suppress the Bcl-2 level and downregulate the expression of Bax, caspase 3, and caspase 9 than NBP+Eda treatment at the dose equivalent to that of 10b.
NF-κB is widely expressed throughout the central nervous system, seen in all cell types and is constitutively expressed in neurons42. The correlation of NF-κB activation with cerebral ischemia was firstly identified in glial cells of postmortem human brains, primarily in the penumbra43. Elevated NF-κB activation has also been reported in various rat models of cerebral ischemia, with initial studies suggesting that this activation promotes cell death. Our data confirm earlier reports of NF-κB activation in the brain after MCAO44 and suggest that 10b can exacerbate the activation of NF-κB seen in the brains of rats after I/R (Figure 7Ba). The data shown in Figure 7Ba indicate that NF-κB was indeed translocated to the nucleus in the 10b-treated rats after I/R, suggesting a role in transcriptional activation. The phosphorylation of the p65 subunit of NF-κB has been shown to increase transcriptional activity (Figure 7Bb)45. It has also been shown that phosphorylation of the p65 subunit is induced in response to a variety of apoptotic stimuli44. The increased phosphorylation of p65 in the 10b-treated rats after I/R suggests that upregulation of NF-κB could be involved in the progression and regulation of the genes that control neuronal apoptosis. To this end, we investigated whether NF-κB activation was correlated with increased IκB-α phosphorylation in our rat model of I/R. Our results clearly demonstrate increased phosphorylation of IκB-α in the NBP-treated and Eda-treated rats with a maximum in those treated with NBP+Eda treatment, which closely resembles the kinetics of phosphorylation of IκB-α in 10b (70 mg/kg) (Figure 7Bd). In the 10b (140 mg/kg) group, the degree of phosphorylation began to return to the levels seen in sham animals and was correlated with the kinetics of NF-κB activation. It thus appears that 10b treatment with an equivalent dose to that used for NBP+Eda demonstrated a greater decrease in the protein levels of the p65 subunit of NF-κB and phosphorylation of IκB-α than the NBP+Eda treatment. In the group treated with 10b at a dose equimolar to NBP+Eda, there was markedly increased expression of phosphorylation of the p65 subunit of NF-κB and IκB-α compared with those treated with NBP+Eda.
We show herein, for the first time, in vivo evidence that NF-κB activation during I/R injury is independent of the proteolytic degradation of IκB-α, and that this regulation is likely to occur at the level of increased tyrosine kinase activity. Its activity is regulated by upregulation of its inhibitor, IκB-α, which can sequester active NF-κB in the cytoplasm46. It is well established that NF-κB can be induced by reactive oxygen species, and, by contrast, antioxidants are capable of inhibiting the proteolytic degradation of IκB47. However, we show here that NF-κB activation is independent of IκB degradation and likely involves phosphorylation of IκB-α, which leads to liberation of NF-κB and subsequent translocation to the nucleus. The finding that NF-κB was not activated by the normal IκB-degradative pathway also ruled out an important role for TNF-α in the initial induction of the immediate-early response genes, because TNF-α-mediated NF-κB activation occurs via IκB-α degradation48. The IκB kinase has yet to be identified, but, based on findings in these studies, this kinase may prove to be an attractive target for early intervention in the inflammatory cascade following the I/R associated with cerebral ischemia. Although the role of NF-κB activation in the context of beneficial effects in the protection against apoptosis has been somewhat controversial, inhibition of NF-κB activation may have an overall beneficial effect for I/R injury in cerebral ischemia.
Conclusion
In the light of current findings, we postulate that compound 10b may play a neuroprotective role by improving mitochondrial function, preventing mitochondria-dependent cell death and promoting the NF-κB/TNF-α signaling pathway. The translocation of Nrf2 to the nucleus, accompanied by upregulation of the endogenous antioxidant systems, was partly responsible for the neuroprotective effect of 10b treatment. In our experiments, 10b (particularly at the higher dose) appears to be more potent in ischemic neuroprotection than NBP, edaravone or the NBP+edaravone mixture. Together, these results indicate that compound 10b is an attractive candidate for intervention of ischemic stroke and merits further study.
Author contribution
Kai HUA and Xiao SHENG performed research, analyzed data and wrote the paper. Ting-ting LI, Lin-na WANG, and Yi-hua ZHANG contributed new reagents and analytic tools. Zhang-jian HUANG and Hui JI designed the research.
References
Margaill I, Plotkine M, Lerouet D . Antioxidant strategies in the treatment of stroke. Free Radic Biol Med 2005; 39: 429–43.
Janardhan V, Qureshi AI . Mechanisms of ischemic brain injury. Curr Cardiol Rep 2004; 6: 117–23.
Crack PJ, Taylor JM . Reactive oxygen species and the modulation of stroke. Free Radic Biol Med 2005; 38: 1433–44.
Duchen MR, Verkhratsky A, Muallem S . Mitochondria and calcium in health and disease. Cell Calcium 2008; 44: 1–5.
Watanabe K, Morinaka Y, Iseki K, Watanabe T, Yuki S, Nishi H . Structure-activity relationship of 3-methyl-1-phenyl-2-pyrazolin-5-one (edaravone). Redox Rep 2003; 8: 151.
Higashi Y, Jitsuiki D, Chayama K, Yoshizumi M . Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Pat Cardiovasc Drug Discov 2006; 1: 85.
Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N . Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev 2006; 12: 9.
Nakamura T, Kuroda Y, Yamashita S, Zhang X, Miyamoto O, Tamiya T, et al. Edaravone attenuates brain edema and neurologic deficits in a rat model of acute intracerebral hemorrhage. Stroke 2008; 39: 463.
Liu C, Liao S, Zeng J, Lin J, Li C, Xie L, et al. dl-3n-Butylphthalide prevents stroke via improvement of cerebral microvessels in RHRSP. J Neuro sci 2007; 260: 106.
Zhu X, Li X, Liu J . Recent pharmacological studies on natural products in China. Eur J Pharmacol 2004; 500: 221.
Yan C, Feng Y, Zhang J . Effects of dl-3-n-butylphthalide on regional cerebral blood flow in right middle cerebral artery occlusion rats. Acta Pharmacol Sin 1998; 19: 117.
Peng Y, Zeng X, Feng Y, Wang X . Antiplatelet and antithrombotic activity of L-3-n-butylphthalide in rats. J Cardiovasc Pharmacol 2004; 43: 876.
Yang CY . Design, synthesis and evaluation of hybrids from edaravone and 3-n-butylphthalide ring-opened derivatives as novel platelet aggregation inhibitors [dissertation]. Nanjing (Jiangsu): China Pharmaceutical University; 2013.
Zhao Q, Zhang C, Wang XL, Chen L, Ji H, Zhang YH . (S)-ZJM-289, a nitric oxide-releasing derivative of 3-n-butylphthalide, protects against ischemic neuronal injury by attenuating mitochondrial dysfunction and associated cell death. Neurochem Int 2012; 60: 134–44.
Longa EZ, Weinstein PR, Carlson S . Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20: 84–91.
Shah ZA, Namiranian K, Klaus J, Kibler K, Dore S . Use of an optimized transient occlusion of the middle cerebral artery protocol for the mouse stroke model. J Stroke Cerebrovasc Dis 2006; 15: 133–8.
Lin TN, He YY, Wu G, Khan M, Hsu CY . Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 1993; 24: 117–21.
Flohe L, Gunzler WA . Assays of glutathione peroxidase. Methods Enzymol 1984; 105: 114–21.
Kakkar P, Das B, Viswanathan PN . A modified spectrophotometric assay of superoxide dismutase. Indian J Biochem Biophys 1984; 21: 130–2.
Rasheed Z, Alzolibani AA, Al-Shobaili HA, Saif GB, Al Robaee AA . Biochemical and immunological studies on erythrocytes superoxide dismutase modified by nitric oxide in patients with alopecia areata: Implications in alopecia patchy persistent and alopecia universalis. Immunol Lett 2014; 160: 50–7.
Mihara M, Uchiyama M . Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem 1978; 86: 271–8.
Song Z, Cui Y, Ding MZ, Jin HX, Gao Y . Protective effects of recombinant human brain natriuretic peptide against LPS-Induced acute lung injury in dogs. Int Immunopharmacol 2013; 17: 508–12.
Thompson JW, Narayanan SV, Perez-Pinzon MA . Redox signaling pathways involved in neuronal ischemic preconditioning. Curr Neuropharmacol 2012; 10: 354–69.
Tang A, Chen X, Lu Q, Zheng N, Wei Y, Wu X . Antihepatotoxic effect of tadehaginoside, extracted from Tadehagi triquetrum (L), against CCl4-lesioned rats through activating the Nrf2 signaling pathway and attenuating the inflammatory response. Inflammation 2014; 37: 1006–14.
Penn JS, Naash MI, Anderson RE . Effect of light history on retinal antioxidants and light damage susceptibility in the rat. Exp Eye Res 1987; 44: 779–88.
Gauntt CD, Ohira A, Honda O, Kigasawa K, Fujimoto T, Masutani H, et al. Mitochondrial induction of adult T cell leukemia derived factor (ADF/hTx) after oxidative stresses in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1994; 35: 2916–23.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997; 275: 1129–32.
Xia X, Su C, Fu J, Zhang P, Jiang X, Xu D, et al. Role of alpha-lipoic acid in LPS/d-GalN induced fulminant hepatic failure in mice: studies on oxidative stress, inflammation and apoptosis. Int Immunopharmacol 2014; 22: 293–302.
Zhu L, Wang J, Wei T, Gao J, He H, Chang X, et al. Effects of naringenin on inflammation in complete freund's adjuvant-induced arthritis by regulating Bax/Bcl-2 balance. Inflammation 2014; 38: 245–51.
Cohen GM . Caspases: the executioners of apoptosis. Biochem J 1997; 326: 1–16.
Karl S, Pritschow Y, Volcic M, Hacker S, Baumann B, Wiesmuller L, et al. Identification of a novel pro-apoptotic function of NF-kappaB in the DNA damage response. J Cell Mol Med 2009; 13: 4239–56.
Chen T, Mou Y, Tan J, Wei L, Qiao Y, Wei T, et al. The protective effect of CDDO-Me on lipopolysaccharide-induced acute lung injury in mice. International immunopharmacology 2015; 25: 55–64.
Lou T, Jiang W, Xu D, Chen T, Fu Y . Inhibitory effects of polydatin on lipopolysaccharide-stimulated RAW 264.7 cells. Inflammation 2015; 38: 1213–20.
Fisher CD, Augustine LM, Maher JM, Nelson DM, Slitt AL, Klaassen CD, et al. Induction of drug-metabolizing enzymes by garlic and allyl sulfide compounds via activation of constitutive androstane receptor and nuclear factor E2-related factor 2. Drug Metab Dispos 2007; 35: 995–1000.
Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB . Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem 2005; 280: 32485–92.
Schulke S, Dreidax D, Malik A, Burmester T, Nevo E, Band M, et al. Living with stress: regulation of antioxidant defense genes in the subterranean, hypoxia-tolerant mole rat, Spalax. Gene 2012; 500: 199–206.
Jaiswal AK . Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 2004; 36: 1199–207.
Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem 1999; 274: 21645–50.
Nishiyama A, Masutani H, Nakamura H, Nishinaka Y, Yodoi J . Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life 2001; 52: 29–33.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166: 193–203.
Zimmermann AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, Linseman DA . Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular basis for antioxidant function at mitochondria. J Biol Chem 2007; 282: 29296–304.
Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA . Constitutive NF-κB activity in neurons. Mol Cell Biol 1994; 14: 3981–92.
Terai K, Matsuo A, McGeer EG, McGeer PL . Enhancement of immunoreactivity for NF-κB in human cerebral infarctions. Brain Res 1996; 739: 343–9.
Shen WH, Zhang CY, Zhang GY . Antioxidants attenuate reperfusion injury after global brain ischemia through inhibiting NF-κB activity in rats. Acta Pharmacol Sin 2003; 24: 1125–30.
Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W . Iκb kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 1999; 274: 30353–6.
Beg AA, Baldwin ASJ . The I kappa B proteins: multifunctional regulators of Rel/NF-kappa B transcription factors. Genes Dev 1993; 7: 2064–70.
Muller JM, Rupec RA, Baeuerle PA . Study of gene regulation by NF-kappa B and AP-1 in response to reactive oxygen intermediates. Methods 1997; 11: 301–12.
Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S . I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell 1995; 80: 573–82.
Acknowledgements
This study was financially supported by grants from the National Natural Science Foundation of China (No 81373419) and the PhD Program Foundation of the Ministry of Education of China, China Pharmaceutical University (No 20130096110005).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Hua, K., Sheng, X., Li, Tt. et al. The edaravone and 3-n-butylphthalide ring-opening derivative 10b effectively attenuates cerebral ischemia injury in rats. Acta Pharmacol Sin 36, 917–927 (2015). https://doi.org/10.1038/aps.2015.31
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.31
Keywords
This article is cited by
-
NADPH is superior to NADH or edaravone in ameliorating metabolic disturbance and brain injury in ischemic stroke
Acta Pharmacologica Sinica (2022)
-
Thioredoxin-Interacting Protein (TXNIP) in Cerebrovascular and Neurodegenerative Diseases: Regulation and Implication
Molecular Neurobiology (2018)
-
14-3-3 Isoforms Differentially Regulate NFκB Signaling in the Brain After Ischemia-Reperfusion
Neurochemical Research (2017)
-
Suppressing Receptor-Interacting Protein 140: a New Sight for Salidroside to Treat Cerebral Ischemia
Molecular Neurobiology (2016)
-
Protective Effect of Astragaloside IV Against Paraquat-Induced Lung Injury in Mice by Suppressing Rho Signaling
Inflammation (2016)
