
Abstract
Aim:
Pharmacodynamic analysis of intravenous recombinant urate oxidase produced by Escherichia coli was performed in healthy subjects using a pharmacokinetic/pharmacodynamic (PK/PD) model.
Methods:
A randomized, single-blind, placebo-controlled study was performed in 40 healthy Chinese subjects (4 groups of 10 subjects each, placebo 4:1 ratio) who received infusions of uricase (single doses of 0.1, 0.2, and 0.3 mg/kg; multiple doses of 0.2 mg·kg−1·d−1 for 7 d). PK profiles were determined through plasma uricase activity, and PD profiles were established using uric acid levels in plasma and urine. The plasma PD parameter was estimated as changes in plasma uric acid levels as the effect in the indirect response model. Adverse events were also monitored.
Results:
A two-compartment PK model with constant iv input and first-order output was used to describe the kinetic process of plasma uricase. The low value (2.8 U/L) of drug concentration that achieved 50% of maximum effect (EC50) indicated that low plasma uricase concentrations were sufficient to produce pharmacological effects. A strong relationship (r2=0.9991) between the mean uric acid concentration in blood and the mean uric acid excretion rate in urine in the range of 11 to 30 h after single dosing was found. Infusions of uricase were well tolerated in all subjects.
Conclusion:
The PK/PD model predicted the effective dose to be 0.1 mg/kg in healthy subjects. The excretion rate of uric acid in urine may be used as a new index for pharmacological effects in further clinical trials.
Similar content being viewed by others


Introduction
Urate oxidase can catalyze uric acid into an inactive and soluble metabolite, allantoin, which is easily excreted by the kidney1. This enzyme exists endogenously in most mammals, but this special elimination of uric acid is inoperative in humans due to a mutation in the uricase gene during evolution. Plasma uric acid rises and accumulates when less uric acid is excreted than produced, which leads to hyperuricemia. Allopurinol, a xanthine oxidase inhibitor, has traditionally been used to treat hyperuricemia. It can inhibit the conversion of hypoxanthine and xanthine to uric acid. However, allopurinol also has its limitations, such as a slow onset of therapeutic effect and adverse reactions to allopurinol2,3.
Rasburicase, a recombinant urate oxidase, has been approved in the United States as an alternative to allopurinol for the management of plasma uric acid levels in pediatric patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anti-cancer therapy expected to result in tumor lysis and a subsequent elevation of plasma uric acid. The approved dosing regimen is 0.15 or 0.2 mg·kg−1·d−1 for 5 d intravenously (iv) over 30 min4. Rasburicase, derived from Aspergillus flavus, is produced by a genetically modified Saccharomyces cerevisiae strain with a long culture period and high price4,5. Therefore, low-cost preparation for uricase seems essential.
Escherichia coli, which is widely used to obtain recombinant proteins, has several advantages over other expression systems, including its low cost, high productivity and well-characterized genetics6. Actually, a new uricase product prepared by E coli has been obtained in our laboratory. The efficacy and pharmacokinetics of this product have been studied, and the dosing regimen is 0.1–0.3 mg/kg7,8. However, the observed efficacy was not directly related to plasma concentrations of the drug based on the known pharmacokinetic (PK) and pharmacodynamic (PD) characteristics of uricase7. Therefore, a PK/PD model might be fitted to predict the pharmacological responses of the plasma uric acid levels. This type of PK/PD model for intravenous uricase has not been reported.
Therefore, the present study was designed primarily to analyze the pharmacodynamics of uricase produced by E coli using a PK/PD model that can describe previously published data obtained in healthy subjects and allow a rational design for dose regimens to support a clinical phase II study. An additional objective was to characterize the safety profiles and immune response to this new uricase product in a Chinese phase I study.
Materials and methods
Ethical statement
This study was performed in accordance with the declaration of Helsinki and its amendments; the Ethical Committee of Xiangya Third Hospital of Central South University approved the experimental protocol. Written informed consent was obtained from all subjects before commencing the study.
Chemicals and reagents
Uric acid was purchased from Sigma-Aldrich (St Louis, MO, USA). 1,3-15N uric acid was purchased from ISOTEC (Great Neck, NY, USA). Both chemicals were of at least 98% purity. Acetonitrile and methanol were of LC-MS grade, and formic acid was of LC grade (Tedia Company Inc, Fairfield, OH, USA). All other chemicals were of AR grade and available from commercial sources.
Recombinant urate oxidase derived from A flavus was expressed in E coli. The urate oxidase was a tetrameric protein that consisted of identical subunits. The average molecular weight of each subunit was 34.2 kDa. Further characterizations were performed using reverse-phase high-performance liquid chromatograph (RP-HPLC), size exclusion chromatography-HPLC (SEC-HPLC), sodium dodecylsulfonate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting, which indicated at least 95% purity9. The uricase product (recombinant urate oxidase for injection) was manufactured by Weihai Anjie Medical and Biological Technology Co, Ltd (Weihai, Shandong province of China) according to Good Manufacturing Practice and was supplied in vials containing 1.5 mg uricase (26.4 EAU) in 1 mL water for injection.
Subjects
Forty volunteers were enrolled in this study. All subjects were in good health, as indicated by their medical history, physical examinations, and routine laboratory tests (hematology, blood chemistries and urinalysis). All subjects were asked to abstain from alcohol, caffeine and grapefruit juice for two weeks before the study. All subjects were non-smokers and were not allowed to use any drugs two weeks before the administration of the study drug. A standardized meal that can not affect the level of uric acid was served to all subjects one week before or during the study. Subjects with G6PDH or cytochrome B5 deficiency were also excluded.
Study design
Details of the clinical protocol have been described previously7,8. Briefly, a randomized, single-blind, placebo-controlled, phase I study was performed in 40 healthy volunteers. In the single-dose study, subjects were sequentially assigned to one of three dose-escalating groups of 0.1, 0.2, and 0.3 mg/kg of 10 subjects each, including 2 placebo, and received single iv infusions of uricase in 50 mL of a 0.9 % sodium chloride solution that lasted 30 min. Blood samples were collected prior to dosing; at 0.167, 0.333, and 0.5 h during dosing; and at 0.5, 1, 2, 4, 6, 10, 14, 24, 36, 48, 60, and 72 h after dosing. Urine samples were obtained at 0- to 72-h time intervals. In the multiple-dose study, subjects were administered a dose of 0.2 mg/kg or placebo once a day for 7 d. On d 5, 6, and 7, blood samples were collected before dosing for determination of trough concentrations at steady state. On d 7, the sampling times were the same as those used in the single-dose study. Urine samples were obtained for each 24-h interval on d 1–6 and an additional 72 h after the last dose. All subjects were kept in a study unit to ensure proper sample collection. All blood samples for uric acid analysis were kept frozen at -20 °C; plasma specimens for uricase activity assessments were stored at 4 °C to prevent a decline of uricase activity. All urine samples were collected without fixed time interval in order to reflect the rapid decrease of urinary uric acid levels. The volume of each urine sample was measured and recorded. The collection time interval and the median time of each interval were also recorded. A total volume of 10 mL of each urine sample was stored at −20 °C until analysis.
Evaluation of safety
The major aim of the placebo group in our clinical phase I study was to evaluate the safety and tolerability of the three dosing groups of 0.1, 0.2, and 0.3 mg/kg. Physical examinations, vital signs, ECGs, and routine laboratory analyzes were performed before dosing and during the 10 d after dosing. Adverse events that were detected by the principal investigator or were spontaneously reported by study subjects were recorded.
Immune response to uricase
The IgG antibody response to uricase was assessed prior to dosing and on d 7, 14, and 28 post-dosing. If a positive response was observed on d 28, subjects were asked to return to the unit for another determination on d 42.
Analytical methods
The activity of urate oxidase
Uricase activity was assessed using a new LC-MS/MS method developed in our laboratory8. The limit of quantification was set at 2.1 U/L. The accuracy and precision at our three concentrations were below 10.6%.
Uric acid in blood or urine and the immune response to uricase
Uric acid in blood was assayed using a method developed in our laboratory7. The limit of quantification was 0.4 mg/L for uric acid. Accuracy and precision at our three concentrations were below 4.5%. In addition, determinations of uric acid in urine with automatic biochemical analyzer (Hitachi, Japan) and enzyme-linked immunosorbent assays (ELISAs) for IgG-antibodies to uricase (Biotek, USA) were performed in the clinical laboratory of Xiangya Third Hospital.
Data analysis
Pharmacokinetic model
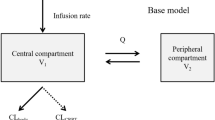
The compartmental pharmacokinetic analysis for each subject was performed using WinNonLin Version 6.1. According to statistical analysis and “goodness-of-fit” criteria, a weighted (1/Y) two-compartment pharmacokinetic model with constant iv input and first-order output was adopted to describe uricase activity in plasma following three single iv doses of 0.1, 0.2, and 0.3 mg/kg.
Pharmacodynamic model
An indirect pharmacological response model using changes in plasma uric acid (ΔCurate, p) as the PD effect was used to examine the PK/PD relationship. The complete model consisted of two parts. First, the PK model described above was used to estimate K10, K12, K21, and V1. Second, an indirect response model with a stimulation of input was fitted to the PD data using the PK parameters obtained in the first step to best explain the relationship between the concentration and effect according to the following equation:

where R is the measured response to intravenous uricase, substituted by ΔCurate,p and ΔCurate,p=|C(urate,p) t=0–C(urate,p) t=t|, Kin is the zero-order constant for the production of response, Kout is the first-order rate constant for loss of response, Cp is the fitted plasma concentration of uricase, Emax means the maximum effect and EC50 means the concentration in plasma that achieves 50% of predicted maximum effect.
Statistical analysis
The best fitted models that described the pharmacokinetic and pharmacodynamic data were selected using statistical analyses and “Goodness-of-fit” criteria, including correlation matrices of the parameters, Akaike information criteria (AIC), and Schwarz Bayes criteria (SBC). The mean uric acid excretion rate in urine was calculated from the amount of urinary excreted uric acid divided by the urine collection interval. Relationships between mean uric acid concentrations in blood and mean uric acid excretion rates in urine were examined using linear regression. All statistical analyses were performed using the SPSS 17.0 statistics software, and P values less than 0.05 were considered significant.
Results
Pharmacokinetic analysis
This study enrolled 40 normal healthy volunteers (20 men, 20 women; age, 22.1±1.9 years; weight, 57.7±6.3 kg; height, 165.6±0.1 cm; body mass index, 21.0±1.4 kg/m2). The main uricase pharmacokinetic parameters are given in Table 18. No sex differences were found in the pharmacokinetic properties of uricase. After multiple dosing, the mean minimum concentrations (Cmin, ss) of uricase on d 5, 6, and 7 were 15.2±3.2, 13.3±2.2, and 13.6±1.8 U/L, respectively. No significant differences between these values were observed. The accumulation index was 1.6±0.2, which suggests that minimal accumulation would appear after multiple doses of uricase. The two-compartment PK model was used to describe the kinetic process of plasma uricase. Plots of the observed and predicted plasma concentration data-time, with the correlations between observed and predicted values, AIC and SBC values of curve fitting, are summarized in Figure 1.

The time course of observed (experimental) and predicted (modeled) values for plasma uricase data according to a two compartment PK model. (A) 0.1 mg/kg group (n=8); (B) 0.2 mg/kg group (n=8); (C) 0.3 mg/kg group (n=8).
Pharmacodynamic analysis
The pharmacodynamic markers of uricase based on uric acid levels in plasma have been reported previously7. No sex differences were observed between these values. The results for the changes in plasma uric acid level versus time profiles at three single doses are shown in Figure 2. Uricase activities (PK data) and ΔCurate, p (PD data) were fitted to the indirect response model. The estimated values of the PD parameters and statistical results for the fitted PD model are reported in Table 2.

The effect data-time curves in healthy Chinese subjects after single infusion doses of uricase (0.1 mg/kg group, n=8; 0.2 mg/kg group, n=8; 0.3 mg/kg group, n=8). Effect: ΔCurate, p=|C(urate, p)t=0−C(urate,p) t=t|.
Relationship between PD biomarkers
Scatter plots of individual uric acid excretion rates in urine vs median collection times after single and multiple doses are presented in Supplementary information Figures 1 and 2, respectively. As a PD biomarker, the urinary excretion rate of uric acid decreased obviously at 12 h after single dosing, with a slow elevation at 36 h. When multiple doses administered, the uricase effect persisted from 12 h after the first dosing to 48 h after the last dosing.
Table 3 shows the values of the mean uric acid concentrations in blood and the mean uric acid excretion rates in urine from 11 to 30 h (the median collection time of urine samples) after single dosing. A strong relationship was found between these two parameters using linear regression (Table 3).
Safety and tolerability evaluation
Intravenous infusions of uricase were well tolerated in all healthy subjects. No serious adverse events (AEs) or clinically important changes in physical examinations were observed among baseline, during the study and at the end of the study. One volunteer in the placebo group was administered anti-inflammatory drugs to treat sore gums. This co-medication did not influence the safety assessment of the study drug by primary investigators.
AEs were observed in 8 (20%) of the 40 subjects: 2 subjects (25%) in the placebo group (urine occult blood positive), 3 subjects (37.5%) in the 0.1 mg/kg group (urine occult blood, ketones and white blood cells positive in 2 of 3; the other subject had elevated serum potassium values), 1 subject (12.5%) in the 0.3 mg/kg group (urine occult blood, ketones and white blood cells positive), and 2 subjects (25%) in the multiple-dose group (urine occult blood and nitrite positive). In addition to abnormal urinalysis in the multiple-dose groups, one of these two subjects had elevated total bilirubin values. However, these changes were not clinically significant.
IgG-antibodies for uricase
No detectable IgG-antibodies were found in the single-dose study on d 7, 14, and 28. Positive results developed in 3 (37.5%) subjects in the multiple-dose study on d 14 and 6 subjects (75%) on d 28. These subjects had negative results for IgG antibodies when they were re-tested on d 42 post-infusion.
Discussion
The classical two-compartment pharmacokinetic model with constant iv input and first-order output was used to describe the disposition kinetics of uricase in humans. The fitted curves of plasma uricase activities in Figure 1 were similar to those obtained in a previous report, in which the pharmacokinetics of iv uricase were dose-proportional over the entire range from 0.1 to 0.3 mg/kg8. Half-life values were approximately 18 h for the three dosing groups. The estimated ratio of AUC(0–t)–multiple to AUC(0–t)–single was 0.9, which indicates a lack of uricase accumulation. These results are also consistent with previous data on the pharmacokinetics of rasburicase in European and North American populations1 and Japanese patients10. Hence, racial differences are not related to the pharmacokinetic properties of uricase protein. Nevertheless, the estimated accumulation index was 1.6 (Table 1), which is greater than the actual accumulation calculated by AUC(0–t)–multiple/AUC(0–t)–single=0.9. This discrepancy may be attributed to the inter-individual variability in the pharmacokinetics of uricase between the two groups.
Figure 2 indicate that iv dosing of 0.3 mg/kg uricase produced much higher responses than observed in the 0.1 mg/kg or 0.2 mg/kg groups, but the responses of the latter doses seemed to be identical. The onset of action was rapid because changes in plasma uric acid levels reached a maximum response between 16–22 h post-infusion (Figure 2). Urine samples after dosing were collected and compared with changes in plasma uric acid levels. The efficacy of uricase was also supported by a decreased urine uric acid excretion rate (Supplementary Figures 1 and 2), which paralleled the observations of plasma uric acid levels at the corresponding median time (data not shown). Unfortunately, no PD model could be established because urine samples were not collected at baseline, but a strong regression relationship (r2=0.9991) between two PD biomarkers (Table 3) suggested that the measurement of urine uric acid excretion rate may provide an alternative assessment to monitor the effect of uricase therapy. This approach has the advantage of not interfering with plasma uricase when uric acid concentrations are determined using ELISA in clinical examinations. Further studies should focus on the establishment of a uricase PD model using urinary data where the excretion rate of uric acid in urine at baseline is needed.
A time-delay existed between the uricase peak concentration and uric acid minimum concentration in the combination of PK and PD7. Therefore, the pharmacological effect required time to develop. An indirect pharmacological response model was used to examine the pharmacokinetic/pharmacodynamic relationships because uricase is already at the site of action following iv administration. Notably, the application of an indirect response model for data analysis requires knowledge of the mechanism of action of the drugs. Changes in the plasma uric acid levels following uricase administration are due to the conversion of uric acid into allantoin, which can be easily excreted by the kidney. This conversion causes the increase in the observed response in plasma (Figure 2). Therefore, an indirect response model with the stimulation of input was considered appropriate for describing our data. The parameter of most interest in the PK/PD model was EC50, which is the plasma uricase concentration that is needed to produce a 50% maximal effect, and had a relatively low value of 2.8 U/L (Table 2). According to the activity–time profiles (Figure 1), the dosing regimens of 0.1, 0.2, and 0.3 mg/kg maintained plasma uricase above the EC50 for most of the time post-infusion, which suggests that these three doses of uricase were equally effective at reducing uric acid concentrations in plasma. These results suggest that a single dose of 0.1 mg/kg may be sufficient to control plasma uric acid.
The results of the safety evaluation suggested that uricase infusion in our population was well tolerated at all doses. No new safety concerns were found for healthy Chinese subjects. The data also indicated no major differences in the frequencies of AEs between the three dosing groups. Antibodies against uricase in our study were detected in 3 (37.5%) of the 8 subjects on d 14 and 6 subjects (75%) on d 28. These numbers seemed to be higher than previously published data on rasburicase where 5 (10%) of the 50 Japanese adult patients with leukemia or lymphoma had anti-uricase antibodies on d 2910. One likely explanation for the apparent difference in immunogenicity compared with rasburicase is that the leukemia patients treated with rasburicase were probably more immunosuppressed because of their disease and the chemotherapy that they were receiving. Antibodies against exogenous proteins would enhance hypersensitivity reactions, and the clinical significance of the development of antibodies should be a major concern. Nevertheless, uricase was not highly immunogenic in our study, although a higher incidence of antibody development was observed. Actually, no subjects experienced hypersensitivity reactions or positive results for IgG antibodies when re-tested on d 42 post infusion.
In conclusion, the developed PK/PD model predicted the effective uricase dose as 0.1 mg/kg in healthy subjects, which promotes the appropriate use of uricase and obviates the need for the use of higher dosages. The uricase product produced by E coli was well tolerated and effective in Chinese subjects. The present work focused on a clinical phase I study, which is the first required step before a multiple-center phase II study can be performed, and our findings provide support for further studies to determine a safe and accurate concentration/effect relationship of this uricase product at one dose of 0.1 mg/kg in therapeutic patient populations. The urine uric acid excretion rate might be a new index for PD response in further clinical trials.
Author contribution
Ning-fang CAI, Ze-neng CHENG, and Xi LUO designed research; Ying ZI, Zhi LIU, and Li-yun ZHENG performed research; Xin GUO and Zhi LIU contributed analytic tools; Ning-fang CAI, Ying ZI, and Xi LUO analyzed data; Ning-fang CAI and Xi LUO wrote the paper.
References
Pui CH . Rasburicase: a potent uricolytic agent. Expert Opin Pharmacother 2002; 3: 433–42.
Greene ML, Fujimoto WY, Seegmiller JE . Urinary xanthine stones — a rare complication of allopurinol therapy. N Engl J Med 1969; 280: 426–7.
Andreoli SP, Clark JH, McGuire WA, Bergstein JM . Purine excretion during tumor lysis in children with acute lymphocytic leukemia receiving allopurinol: relationship to acute renal failure. J Pediatric 1986; 109: 292–8.
Liu CY, Sims-McCallum RP, Schiffer CA . A single dose of rasburicase is sufficient for the treatment of hyperuricemia in patients receiving chemotherapy. Leukemia Res 2005; 29: 463–5.
Leplatois P, Le Douarin B, Loison G . High-level production of a peroxisomal enzyme: aspergillus flavus uricase accumulates intracellularly and is active in Saccharomyces cerevisiae. Gene 1992; 122: 139–45.
Legoux R, Delpech B, Dumont X, Guillemot JC, Ramond P, Shire D, et al. Cloning and expression in Escherichia coli of the gene encoding Aspergillus flavus urate oxidase. J Biol Chem 1992; 267: 8565–70.
Luo X, Cai N, Cheng Z . Determination of uric acid in plasma by LC-MS/MS and its application to an efficacy evaluation of recombinant urate oxidase. Anal Sci 2013; 29: 709–13.
Luo X, Cai NF, Cheng ZN . Development of a new LC-MS/MS based enzyme activity assay for recombinant urate oxidase in plasma and its application to pharmacokinetics in human. J Pharm Biomed Anal 2013; 81–82: 8–12.
Zhu L, Wang QM, Wu GD, Xue TT, Sun LX, Wang JY . Expression, purification and activity identification of urate oxidase in Escherichia coli. China Biotechnol 2011; 31: 83–6.
Ishizawa K, Ogura M, Hamaguchi M, Hotta T, Ohnishi K, Sasaki T, et al. Safety and efficacy of rasburicase (SR29142) in a Japanese phase II study. Cancer Sci 2009; 100: 357–62.
Acknowledgements
The research was founded by the National Natural Science Foundation of China (No 81072700). In addition, we gratefully thank Shuang YANG, An YAO, and Lan-ni LI for gathering the biological samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
(Supplementary figures are available at Acta Pharmacologica Sinica's website.
Supplementary information
PowerPoint slides
Rights and permissions
About this article

Cite this article
Cai, Nf., Cheng, Zn., Zi, Y. et al. Pharmacodynamic analysis of intravenous recombinant urate oxidase using an indirect pharmacological response model in healthy subjects. Acta Pharmacol Sin 35, 1447–1452 (2014). https://doi.org/10.1038/aps.2014.81
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.81
Keywords
This article is cited by
-
Pharmacokinetics of Polyethylene Glycol-Modified Canine Uricase Following Single and Multiple Intravenous Injections in Cynomolgus Monkeys
European Journal of Drug Metabolism and Pharmacokinetics (2020)

{kind=link}
{kind=link}