
Abstract
Aim:
Proteinuria is not only a common marker of renal disease, but also involved in renal tubulointerstitial inflammation and fibrosis. The aim of this study was to investigate the mechanisms underlying the protective effects of enalapril, an ACEI, against nephropathy in rats.
Methods:
Wistar rats underwent unilateral right nephrectomy, and then were treated with BSA (5 g·kg−1·d−1, ip), or BSA plus enalapril (0.5 g·kg−1·d−1, po) for 9 weeks. The renal lesions were evaluated using histology and immunohistochemistry. The expression of NLRP3, caspase-1, IL-1β and IL-18 was analyzed using immunohistochemistry, RT-PCR and Western blot.
Results:
BSA-overload resulted in severe proteinuria, which peaked at week 7, and interstitial inflammation with prominent infiltration of CD68+ cells (macrophages) and CD3+ cells (T lymphocytes), particularly of CD20+ cells (B lymphocytes). BSA-overload markedly increased the expression of NLRP3, caspase-1, IL-1β and IL-18 in the proximal tubular epithelial cells, and in inflammatory cells as well. Furthermore, the expression of IL-1β or IL-18 was significantly correlated with proteinuria (IL-1β: r=0.757; IL-18: r=0.834). These abnormalities in BSA-overload rats were significantly attenuated by concurrent administration of enalapril.
Conclusion:
Enalapril exerts protective effects against BSA-overload nephropathy in rats via suppressing NLRP3 inflammasome expression and tubulointerstitial inflammation.
Similar content being viewed by others

Introduction
Proteinuria is a common clinical manifestation of renal disease, especially in patients with chronic kidney disease (CKD)1. Experimental evidence demonstrates that proteinuria can arise from glomerular and tubular diseases and is an important renal toxin2. The presence and severity of proteinuria is correlated with the prognosis of patients with CKD, largely because proteinuria accelerates CKD progression to end-stage renal failure by stimulating tubulointerstitial inflammation and fibrosis3. Therefore, it is widely recognized that control of proteinuria is a key strategy for treating CKD.
Angiotensin-converting enzyme inhibitors (ACEIs) have been studied within a variety of CKDs4 and are considered the most effective therapies for delaying the progression of CKD by reducing hypertension and proteinuria and subsequently halting renal fibrosis5. Recently, Radaelli et al reported that enalapril and ramipril prevent systemic inflammatory responses during on-pump coronary artery bypass grafting6. Other research has suggested that XJP-1, a novel ACEI, is able to inhibit the inflammatory response of HUVECs7. However, the underlying molecular mechanisms by which ACEIs are able to protect against CKD remain elusive.
The innate immune system is able to recognize and respond to foreign molecular structures as well as abnormal self-molecules. The inflammasome is an important component of the innate immune response against a broad range of exogenous and endogenous danger signals8. Inflammasomes are a group of multimeric protein complexes that consist of an inflammasome sensor molecule, the adaptor protein ASC, and caspase-1. Once the protein complexes have formed, inflammasomes sense microbial molecules and danger signals in the cytoplasm of cells and activate caspase-1 in response. Caspase-1 activation causes the proteolytic activation of the pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18)9. Of the NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is the most characterized inflammasome and has been shown to play a key role in regulating the immune response within a variety of diseases10,11.
The NLRP3 inflammasome can be activated by a wide range of danger signals, including both xenobiotics12 and abnormal self-molecules resulting from metabolic dysregulation13. Recently, many studies have demonstrated the role of the inflammasome in the pathogenesis of renal disease14. Vilaysane et al suggested activation of the NLRP3 inflammasome in unilateral ureteral obstruction (UUO) mice15. Mulay et al demonstrated that CaOx crystals triggered IL-1β secretion via the NLRP3/ASC/caspase-1 axis16. Correa-Costa et al reported NLRP3 gene expression in tubulointerstitial nephritis induced by adenine-enriched food17. Previously, we have demonstrated that high doses of BSA activate renin angiotensin system (RAS) in HK2 cells18. Furthermore, we demonstrated that BSA could directly activate the NLRP3 inflammasome in HK2 cells19. Therefore, we hypothesized that activation of the NLRP3 inflammasome might be associated with intrarenal RAS activation and that ACEIs exert their reno-protective role by inhibiting the NLRP3 inflammasome and tubulointerstitial inflammation.
Materials and methods
Animals and experimental design
Healthy 6- to 8-week-old male Wistar rats weighing 150 to 190 g were purchased from The Academy of Military Medical Laboratory Animal Center (Beijing, China). Animal care and treatment were conducted in accordance with the approval of the Ethical Committee of Southeast University. All animals were housed in a room in which the temperature was kept constant on a 12-h light/dark cycle at 22±3 °C and fed a standard diet. All animals underwent unilateral right nephrectomy five days before the initiation of intraperitoneal injections, and the rats were randomly assigned into three groups: (1) the BSA-overload group (n=8) received daily intraperitoneal injection of 5 g/kg bovine serum albumin (BSA No A-8806, 98%–99% albumin; Sigma Chemical Company, St Louis, MO, USA) and intragastric administration of an equivalent volume of 0.9% saline as enalapril treated group; (2) the enalapril treated group (n=10) simultaneously received daily intraperitoneal injection of 5 g/kg BSA and intragastric administration of 0.5 g/kg enalapril (dissolved in 0.9% saline, Roche Group, Switzerland) after 1 week of intraperitoneal injections of BSA; and (3) the control group (n=10) received daily intraperitoneal injection and intragastric administration of an equivalent volume of 0.9% saline. After 9 weeks, the animals were anesthetized with pentobarbital sodium (150–200 mg/kg, intraperitoneally). Blood was collected, and the kidneys were perfused with cold sodium saline and removed.
Plasma and urine determinations
Animals were housed individually in metabolic cages, and 24-h urine samples were collected in metabolic cages both before injections and at weeks 2, 5, 7, and 9. A biuret assay method was used to determine the 24-h urinary protein. Plasma was obtained by exsanguination through the abdominal aorta, and blood total protein (TP), albumin (ALB), serum creatinine (Scr) and blood urea nitrogen (BUN) were determined by automatic analyzers (Hitachi, Tokyo, Japan).
Histopathological examination
For light microscopy, the renal tissue was fixed in 10% buffered formalin and embedded in paraffin, and sections (3 μm) were stained with Periodic Acid-Schiff (PAS) and Masson's trichrome to evaluate histopathological injury. The extent (percentage) of tubulointerstitial injury (inflammatory cells, tubular dilatation/atrophy, interstitial fibrosis, and tubular cast formation) was scored in each tissue section, and the lesions were graded from 0 to 3 (0, normal; 1, lesions involving <25% of the cortical area; 2, 25%–50%; and 3, >50%), as described previously20. The score index in each rat was expressed as the mean value of all scores obtained. Histological parameters were scored by light microscopy on 10 randomly selected nonoverlapping specimens from each rat at 200×magnification on PAS and Masson's trichrome staining. All pathological sections were evaluated by 2 investigators in a blinded manner.
Immunohistochemical staining analysis
Formalin-fixed, paraffin-embedded samples were deparaffinized and hydrated through decreasing concentrations of ethanol. Heat-induced antigen retrieval was conducted in 10 mmol/L sodium citrate solution (pH 6.0) for 15 min in a microwave. Slides were washed three times in PBS. Primary antibodies against NLRP3, caspase-1, IL-1β, IL-18, CD3, CD20, and CD68 (Santa Cruz Biotechnology, Santa Cruz, USA) were diluted to the manufacturers' recommended concentrations and applied for overnight incubation at 4 °C. A streptavidin peroxidase detection system (Maixin Biotechnology Co Ltd, Hong Kong, China) was used according to the manufacturer's protocol. Subsequently, the sections were stained with diaminobenzidine and counterstained with hematoxylin. The semiquantitative immunohistochemical analysis was scored using Image-Pro plus 6.0 software in ten randomly selected cortical sections in each tissue section.
Western blot analysis
The remaining tissue was frozen with liquid nitrogen and stored at −80 °C for Western blot and real-time PCR. Briefly, the total proteins extracted from the renal cortex were separated by SDS PAGE and then transferred onto a nitrocellulose membrane. Nonspecific binding to the membrane was blocked by a preincubation of the membranes in 1×TBS containing 5% skim milk for 1 h at room temperature. The membranes were then incubated overnight at 4 °C with the primary antibodies against NLRP3, caspase-1, IL-1β, and IL-18. Binding of the primary antibodies was followed by incubation for 1 h at room temperature with the appropriate peroxidase-conjugated secondary antibodies. The signals were visualized with the enhanced chemiluminescence system (GE Healthcare, Chalfont St Giles, UK) and captured on X-ray film. Quantification of Western blot results was performed using Image J software (NIH, Bethesda, Maryland, USA), as described previously21.
Real-time PCR analysis
Total RNA was extracted from the kidney using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. cDNA was synthesized using a reverse transcription system kit (Takara, Dalian, China). Real-time quantitative PCR was performed on an ABI PRISM 7300 real-time PCR System (Applied Biosystems, Foster City, CA, USA), and β-actin was served as a control for target genes for reaction efficiency. Results were analyzed using the comparative cycle threshold method22.
Statistical analysis
All results were expressed as the mean±SD. Each experiment was repeated three times independently. Data were analyzed using either the t test or ANOVA where appropriate using SPSS 17.0 software, with P<0.05 regarded as statistically significant.
Results
Changes in physical and biochemical parameters in experimental animals
Proteinuria was detectable at week 2 after intraperitoneal injections of BSA in both the BSA-overload and enalapril groups (P<0.05; compared with control group). During the experimental time course, BSA-overload animals developed significant proteinuria, which peaked at week 7. However, the enalapril-treated group exhibited significantly reduced proteinuria compared with the BSA-overload group (P<0.05). No significant differences in body weight, TP, serum albumin, or Scr were observed among the three groups. BSA-overload animals exhibited a higher BUN level than the control animals (Figure 1).

Changes of physical and biochemical parameters in experimental animals. (A) There is no significant difference in the body weight between the groups. (B) BSA-overload animals developed significant proteinuria, which peaked at week 7, and enalapril treatment could significantly reduce the proteinuria. (C and D) There were no significant differences among the groups in total protein (TP) or albumin (ALB) or creatinine (Scr). (E) BUN was significantly higher in BSA-overload animals compared to its control. Data are represented as mean±SD. n=3 experiments. bP<0.05 vs control group. eP<0.05 vs BSA-overload group.
Enalapril attenuated proximal tubular cell damage in BSA-overload rats
In the BSA-overload animals, protein droplets accumulated in the cytoplasm of the proximal tubular cells. Changes of the tubular architecture included tubular dilatation and vacuolization, atrophy, proteinaceous casts in the proximal and distal tubules, and interstitial inflammation with heavy macrophage and lymphocyte infiltration. Only mild glomerular mesangial expansion was observed in this BSA-overload rat model (Figure 2A). Enalapril treatment significantly attenuated the above morphological changes. Semiquantitative analysis revealed there was a significant increase in the tubulointerstitial injury score in the BSA-overload group compared with the control group (P<0.05), which was significantly attenuated by treatment with enalapril (P<0.05; Figure 2B).

Enalapril attenuated proximal tubular cell damage. (A) PAS and Masson stains of tissue as shown (×200). Tubular dilatation and vacuolization, atrophy, proteinaceous casts in the proximal and distal tubules, and interstitial inflammation with heavy macrophage and lymphocyte infiltration were observed in the BSA-overload animals. Enalapril attenuated these changes. (B) Tubulointerstitial injury score. The BSA-overload group exhibited a significant increase in the tubulointerstitial injury score, and enalapril decreased the score. Data are represented as mean±SD. n=3 experiments. bP<0.05 vs control group. eP<0.05 vs BSA-overload group.
Enalapril inhibited the interstitial inflammation in BSA-overload rats
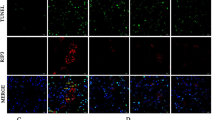
BSA overload induced a prominent infiltration of inflammatory cells into the renal interstitial area; most of them were CD3+ (T lymphocytes), CD20+ (B lymphocytes), and CD68+ cells (macrophages) (Figure 3A). After treatment with enalapril, all types of tubulointerstitial infiltration inflammatory cells significantly decreased in number (P<0.05; Figure 3B).

Enalapril exerted anti-inflammatory effects. (A) Immunohistochemical staining of tissue as shown (×200). BSA overload induced a prominent infiltration of CD3+ (T lymphocytes), CD20+ (B lymphocytes), and CD68+ cells (macrophages) into the renal interstitial area, and enalapril decreased the inflammatory cells. (B) The number of T cells, B cells, and macrophages in the three groups; there were more CD20+ cells than other cell types. Data are represented as mean±SD. n=3 experiments. bP<0.05 vs control group. eP<0.05 vs BSA-overload group.
Influence of enalapril on NLRP3 inflammasome activation
To determine whether the infiltration of inflammatory cells was associated with inflammasome activation in BSA-overload-induced nephropathy, we determined the intensity and distribution of NLRP3, caspase-1, IL-1β, and IL-18 as markers of inflammasome activation. Immunohistochemical staining indicated that NLRP3, caspase-1, IL-1β, and IL-18 were not only expressed in inflammatory cells but also in proximal tubules (Figure 4A). More importantly, NLRP3, caspase-1, IL-1β, and IL-18 expression in the BSA-overload group was significantly increased compared with that in the control group (P<0.05; Figure 4B). Furthermore, we found that there were significant correlations between proteinuria and IL-1β/IL-18 expression (r=0.757, P<0.05 for IL-1β; r=0.834, P<0.05 for IL-18; Figure 4C).

Enalapril inhibited NLRP3 inflammasome activation. (A) Representative examples of immunostaining for NLRP3, caspase-1, IL-1β, and IL-18 (×200). NLRP3, caspase-1, IL-1β, and IL-18 were expressed in inflammatory cells as well as in proximal tubules, and enalapril inhibited NLRP3, caspase-1, IL-1β, and IL-18 expression. (B) Semiquantitative analysis using Image-Pro plus 6.0 software. Data are represented as mean±SD. n=3 experiments. bP<0.05 vs control group. eP<0.05 vs enalapril group. (C) Correlation between proteinuria and expression of IL-1β (a) and IL-18 (b) in BSA-overload rats. Data are compared using Spearmen correlation coefficients, P<0.05.
The differences in NLRP3, caspase-1, IL-1β, and IL-18 expressions between the three groups were confirmed by protein and mRNA analysis as determined by Western blot analysis (Figure 5A) and real-time PCR (Figure 6), respectively. In the BSA-overload group, NLRP3, caspase-1, IL-1β, and IL-18 protein and mRNA were dramatically upregulated, as was the pro-caspase-1, pro-IL-1β, and pro-IL-18 protein level, and treatment with enalapril significantly ameliorated the expression of these cytokines (P<0.05; Figure 5B, 6).

Influence of enalapril on the expression of NLRP3, caspase-1, IL-1β, and IL-18 by Western blot. Rats were given overload BSA with or without enalapril, and expression of NLRP3, caspase-1, IL-1β, and IL-18 proteins were measured by Western blot (A). Semiquantitative analysis using Image J software. In the BSA-overload group, NLRP3, caspase-1, IL-1β, and IL-18 protein were significantly elevated, as were pro-caspase-1, pro-IL-1β, and pro-IL-18; and enalapril treatment decreased their expression (B). Data are represented as mean±SD. n=3. bP<0.05 vs control group. eP<0.05 vs enalapril group.

Influence of enalapril on the mRNA expression of NLRP3, caspase-1, IL-1β and IL-18 by real-time PCR. BSA-overload treatment significantly induced the increasing expression of NLRP3, caspase-1, IL-1β, and IL-18 at mRNA levels, which were markedly suppressed by the treatment with enalapril. Data are represented as mean±SD. n=3 experiments. bP<0.05 vs control group. eP<0.05 vs enalapril group.
Discussion
In this study, we demonstrated that the NLRP3 inflammasome could be activated in the BSA-overload-induced nephropathy model, accompanied with significant interstitial inflammatory cell infiltration and heavy proteinuria. More interestingly, we found that enalapril, a commonly used ACEI, could significantly attenuate interstitial inflammation as well as the activation of the NLRP3 inflammasome.
Tubular toxicity associated with proteinuria has been described previously23, and it has been documented that persistent proteinuria can directly induce kidney injury, even when the primary insult to the kidney has ceased24. A number of studies have demonstrated that proteinuria induces the expression of many proinflammatory and profibrotic factors, including chemokines, cytokines, monocyte chemotactic proteins, growth factors, and complement activation, which leads to tubulointerstitial inflammation and subsequent fibrosis25. Our previous studies found that albumin induces the activation of the intrarenal RAS through the upregulation of ACE and the downregulation of ACE2 in HK2 cells18. A range of clinical and experimental evidence has suggested that Ang II is the major effector molecule of RAS, playing a key role in proteinuric nephropathy by stimulating the secretion of cytokines and chemokines, thus promoting the infiltration of monocytes and macrophages, although the exact mechanisms are still largely assumptive26. In the present study, we demonstrated that BSA overload could induce proteinuria and significant tubulointerstitial inflammation characterized by infiltration of CD20+ (B lymphocytes), CD3+ (T lymphocytes), and CD68+ cells (macrophages). Furthermore, we observed significantly increased expression of the NLRP3 inflammasome, caspase-1 and IL-1β and IL-18. More interestingly, we found that treatment with enalapril could significantly inhibit the expression of the NLRP3 inflammasome as well as tubulointerstitial inflammation.
The NLRP3 inflammasome belongs to the NOD-like receptor (NLR) family and consists of the NLRP3 scaffold, the ASC (encoded by PYCARD, which is common to all inflammasomes) adaptor, and caspase-113,27. Once activated, NLRP3 undergoes self-oligomerization, which recruits the adaptor protein ASC. Subsequently, ASC oligomerizes, leading to a very large supramolecular structure called the pyroptosome. The pyroptosome recruits and activates procaspase-1 to its activated form, caspase-1, leading to the cleavage of pro-IL-1β and pro-IL-18 into the active pro-inflammatory cytokines IL-1β and IL-1828. Pathogen-associated molecular patterns (PAMPs)29, damage-associated molecular patterns (DAMPs)30, and environmental irritants can all activate NLRP331.
Recently, the focus on the role of the NLRP3 inflammasome in kidney diseases has been raised. Mulay et al demonstrated that CaOx crystals induced renal inflammation by NLRP3-mediated IL-1β secretion in intrarenal mononuclear phagocytes16. Although the NLRP3 inflammasome was firstly characterized in immune cells, recent studies suggest that intrinsic renal cells might be involved in the activation of the NLRP3 inflammasome as well. Vilaysane et al documented that there was increased NLRP3 mRNA/protein expression in UUO mice as well as in tubular epithelial cells (TECs)15. Fang et al reported that proteinuria could induce NLRP3 inflammasome activation in TECs through endoplasmic reticulum stress32. All of the studies suggested that the activation of the NLRP3 inflammasome might be an important mechanism for the development of tubulointerstitial inflammation irrespective of the initial cause. It therefore reasonably assumed that the inhibition of the NLRP3 inflammasome will be a novel strategy for the treatment of CKD.
In this study, we demonstrated that enalapril could significantly attenuate the tubulointerstitial expression of the NLRP3 inflammasome and interstitial inflammation in addition to its reduction of proteinuria in a BSA-overload-induced nephropathy model. The antiproteinuric effect of ACEI has been well studied previously, and this class of drug has been recommened as the first choice of treatment for proteinuric nephropathy according to the KDIGO guideline. However, whether ACEIs is able to inhibit tubulointerstitial inflammation and its related mechanism remains uncertain. Our study here may provide novel insight for ACEIs in treating CKD.
In summary, this study demonstrated that enalapril might play an important renoprotective effect in a proteinuric nephropathy model through the reduction of proteinuria as well as through the inhibition of the activation of the NLRP3 inflammasome and subsequent tubulointerstitial inflammation.
Author contribution
Li-hong DING and Bi-cheng LIU conceived the study and designed the experiments; Li-hong DING, Dan LIU, and Min XU performed experiments; Li-hong DING, Dan LIU, Min XU, Hong LIU, Min WU, Ri-ning TANG, Lin-li LV, Kun-ling MA, and Bi-cheng LIU analyzed data and interpreted the results of experiments; Li-hong DING, Dan LIU, and Min XU prepared figures; Li-hong DING drafted the manuscript; and Bi-cheng LIU designed the study, helped to interpret the results and approved the final version of the manuscript.
References
D'Amico G, Bazzi C . Pathophysiology of proteinuria. Kidney Int 2003; 63: 809–25.
Zoja C, Morigi M, Remuzzi G . Proteinuria and phenotypic change of proximal tubular cells. J Am Soc Nephrol 2003; 14 Suppl 1: S36–41.
Zandi-Nejad K, Eddy AA, Glassock RJ, Brenner BM . Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl 2004; (92): S76–89.
How HY, Sibai BM . Use of angiotensin-converting enzyme inhibitors in patients with diabetic nephropathy. J Matern Fetal Neonatal Med 2002; 12: 402–7.
Baltatzi M, Savopoulos C, Hatzitolios A . Role of angiotensin converting enzyme inhibitors and angiotensin receptor blockers in hypertension of chronic kidney disease and renoprotection. Study results. Hippokratia 2011; 15: 27–32.
Radaelli A, Loardi C, Cazzaniga M, Balestri G, DeCarlini C, Cerrito MG, et al. Inflammatory activation during coronary artery surgery and its dose-dependent modulation by statin/ACE-inhibitor combination. Arterioscler Thromb Vasc Biol 2007; 27: 2750–5.
Fu R, Chen Z, Wang Q, Guo Q, Xu J, Wu X . XJP-1, a novel ACEI, with anti-inflammatory properties in HUVECs. Atherosclerosis 2011; 219: 40–8.
Ogura Y, Sutterwala FS, Flavell RA . The inflammasome: first line of the immune response to cell stress. Cell 2006; 126: 659–62.
Petrilli V, Papin S, Tschopp J . The inflammasome. Curr Biol 2005; 15: R581.
Tan MS, Yu JT, Jiang T, Zhu XC, Tan L . The NLRP3 Inflammasome in Alzheimer's disease. Mol Neurobiol 2013; 48: 875–82.
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation and fibrosis. Hepatology 2014; 59: 898–910.
Martinon F, Agostini L, Meylan E, Tschopp J . Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol 2004; 14: 1929–34.
Schroder K, Zhou R, Tschopp J . The NLRP3 inflammasome: a sensor for metabolic danger? Science 2010; 327: 296–300.
Anders HJ, Muruve DA . The inflammasomes in kidney disease. J Am Soc Nephrol 2011; 22: 1007–18.
Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 2010; 21: 1732–44.
Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest 2013; 123: 236–46.
Correa-Costa M, Braga TT, Semedo P, Hayashida CY, Bechara LR, Elias RM, et al. Pivotal role of Toll-like receptors 2 and 4, its adaptor molecule MyD88, and inflammasome complex in experimental tubule-interstitial nephritis. PLoS One 2011; 6: e29004.
Liu BC, Gao J, Li Q, Xu LM . Albumin caused the increasing production of angiotensin II due to the dysregulation of ACE/ACE2 expression in HK2 cells. Clin Chim Acta 2009; 403: 23–30.
Ding LH, Liu D, Xu M, Liu BC . Expression of NLRP3 inflammasome in the BSA-overloaded rats kidney. Chin J Nephrol 2014; 30: In press.
Pan MM, Zhang MH, Ni HF, Chen JF, Xu M, Phillips AO, et al. Inhibition of TGF-beta1/Smad signal pathway is involved in the effect of Cordyceps sinensis against renal fibrosis in 5/6 nephrectomy rats. Food Chem Toxicol 2013; 58: 487–94.
Dai HY, Zheng M, Tang RN, Ma KL, Ni J, Liu BC . Inhibition of integrin-linked kinase by angiotensin II receptor antagonist, irbesartan attenuates podocyte injury in diabetic rats. Chin Med J (Engl) 2012; 125: 888–93.
Chen JF, Ni HF, Pan MM, Liu H, Xu M, Zhang MH, et al. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am J Physiol Renal Physiol 2013; 304: F676–85.
Baines RJ, Brunskill NJ . Tubular toxicity of proteinuria. Nat Rev Nephrol 2011; 7: 177–80.
Kruger D . The pathophysiology, diagnosis, and management of proteinuria. JAAPA 2012; 25: 32–7.
Wu CC, Chen JS, Lu KC, Chen CC, Lin SH, Chu P, et al. Aberrant cytokines/chemokines production correlate with proteinuria in patients with overt diabetic nephropathy. Clin Chim Acta 2010; 411: 700–4.
Cao Z, Cooper ME . Role of angiotensin II in tubulointerstitial injury. Semin Nephrol 2001; 21: 554–62.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–91.
Haneklaus M, O'Neill LA, Coll RC . Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 2013; 25: 40–5.
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J . Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320: 674–7.
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008; 452: 103–7.
Latz E, Xiao TS, Stutz A . Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411.
Fang L, Xie D, Wu X, Cao H, Su W, Yang J . Involvement of endoplasmic reticulum stress in albuminuria induced inflammasome activation in renal proximal tubular cells. PLoS One 2013; 8: e72344.
Acknowledgements
This study was supported by grants from the Key Project of the National Natural Science Foundation (No 81130010), National Key Basic Research Project (“973”) (No 2012CB517700), and Natural Science Foundation of Jiangsu Province (No BK201106; all to Bi-cheng LIU, Principle Investigator).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ding, Lh., Liu, D., Xu, M. et al. Enalapril inhibits tubulointerstitial inflammation and NLRP3 inflammasome expression in BSA-overload nephropathy of rats. Acta Pharmacol Sin 35, 1293–1301 (2014). https://doi.org/10.1038/aps.2014.66
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.66
Keywords
This article is cited by
-
Caspase-11 promotes renal fibrosis by stimulating IL-1β maturation via activating caspase-1
Acta Pharmacologica Sinica (2019)
-
Dietary iron restriction alleviates renal tubulointerstitial injury induced by protein overload in mice
Scientific Reports (2017)
-
Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro
Acta Pharmacologica Sinica (2015)
-
FTY720 inhibits tubulointerstitial inflammation in albumin overload-induced nephropathy of rats via the Sphk1 pathway
Acta Pharmacologica Sinica (2014)
