
Abstract
Aim:
(−)-Epigallocatechin-3-gallate (EGCG) is one of the most abundant polyphenols in green tea with strong antioxidant activity and various therapeutic effects. In this study, we investigated the anti-fibrotic effects of EGCG and underlying mechanisms in bile duct-ligated (BDL) rats and a liver fibrosis model in vitro.
Methods:
BDL rats were treated with EGCG (25 mg·kg−1·d−1, po) for 14 d, and then the serum, bile and liver samples were collected. Liver fibrosis was assessed by serum, urine and bile biochemistry analyses and morphological studies of liver tissues. TGF-β1-stimulated human hepatic stellate LX-2 cells were used as a liver fibrosis model in vitro. The expression of liver fibrogenic genes and signaling proteins in the PI3K/Akt/Smad pathway was examined using Western blotting and/or real-time PCR.
Results:
In BDL rats, EGCG treatment significantly ameliorates liver necrosis, inflammation and fibrosis, and suppressed expression of the genes associated with liver inflammation and fibrogenesis, including TNF-α, IL-1β, TGF-β1, MMP-9, α-SMA, and COL1A1. In LX-2 cells, application of EGCG (10, 25 μmol/L) dose-dependently suppressed TGF-β1-stimulated expression of COL1A1, MMP-2, MMP-9, TGF-β1, TIMP1, and α-SMA. Furthermore, EGCG significantly suppressed the phosphorylation of Smad2/3 and Akt in the livers of BDL rats and in TGF-β1-stimulated LX-2 cells. Application of LY294002, a specific inhibitor of PI3K, produced similar effects as EGCG did in TGF-β1-stimulated LX-2 cells, but co-application of EGCG and LY294002 did not produce additive effects.
Conclusion:
EGCG exerts anti-fibrotic effects in BDL rats and TGF-β1-stimulated LX-2 cells in vitro via inhibiting the PI3K/Akt/Smad pathway.
Similar content being viewed by others

Introduction
Liver fibrosis is a histological hallmark of liver injury. It is characterized by the excessive deposition of extracellular matrix (ECM) proteins, including collagen1. Hepatic stellate cells (HSCs) are the major source of liver fibrogenesis. Under normal physiological conditions, HSCs remain quiescent. However, when liver cells are injured, inflammatory cytokines and chemokines, including TNF-α and TGF-β, are released by the injured cells and immune cells. TGF-β, in turn, activates HSCs, stimulates ECM deposition, and initiates fibrogenesis2.
A variety of pathogens and conditions can trigger liver fibrogenesis, including cholestasis, hepatic viral infection, nonalcoholic steatohepatitis, and drug toxicity. Persistent liver fibrosis leads to cirrhosis, hepatoma, and/or liver failure. Currently, there is no drug used specifically for treating liver fibrosis or several of its causes, such as cholestasis. Although ursodeoxycholic acid (UDCA) is an FDA-approved medicine for the treatment of primary biliary cirrhosis and intrahepatic cholestasis during pregnancy, it has been demonstrated to have very limited benefits. In fact, a high dose of UDCA has been shown to lead to adverse effects in patients with primary sclerosing cholangitis. Therefore, there is an urgent need for an effective treatment for cholestasis and subsequent liver fibrosis3,4.
(−)-Epigallocatechin-3-gallate (EGCG) is one of the most abundant polyphenols present in green tea. It is not only a strong antioxidant but also a multi-target therapeutic agent. EGCG has been demonstrated to have various beneficial effects in patients5 with and in animal models of cancer, diabetes, viral diseases, Parkinson's disease, Alzheimer's disease, stroke, and obesity6. In addition, recent studies have shown that EGCG reduces liver fibrosis in CCl4-treated rats7 and mice8 and in rats with non-alcoholic fatty liver disease (NAFLD)9. Detailed molecular mechanistic studies have indicated that EGCG can regulate multiple signal transduction pathways in vitro in HSCs10,11,12,13,14, including those involving PDGF-BB, Rho, glutathione synthesis, collagen production and collagenase activity. In particular, Shim et al15 have identified Ras-GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) as a direct target of EGCG in lung cancer cells, in which EGCG suppresses cancer growth.
However, it is not known whether EGCG has any anti-fibrotic effects on cholestatic liver injury. In this report, we assessed the therapeutic effect of EGCG in bile duct-ligated (BDL) rats. We also verified EGCG's antifibrotic effects by assessing collagen expression in a human hepatic stellate cell line, LX-2. Further assessments revealed that the anti-fibrotic effects of EGCG were mainly mediated through the PI3K/Akt/Smad signaling pathway.
Materials and methods
Chemicals and reagents
TGF-β1 (240-B) was purchased from R&D Systems (Minneapolis, MN). EGCG, LY294002, and antibodies against α-smooth muscle actin and β-actin were purchased from Sigma (St Louis, MO, USA). An anti-TGF-β1/2 antibody was purchased from Epitomics (Burlingame, CA, USA). All other antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Real-time PCR master mix was purchased from Roche (Indianapolis, IN, USA). ABI TaqMan primers/probes were purchased from Applied Biosystems (Foster City, CA, USA).
Animal experiments
Male Sprague-Dawley rats (200–230 g) were obtained from the Institute for Experimental Animals, Chinese Academy of Medical Sciences and Peking Union Medical College. The study protocols were in accordance with the regulations of Good Laboratory Practice for non-clinical laboratory studies of drugs issued by the National Scientific and Technologic Committee of People's Republic of China. A total of twenty rats underwent BDL. At 24 h after surgery, the BDL animals were randomly divided into two groups and received daily gavages with either saline or 25 mg/kg EGCG for 14 d. As a healthy control, we also performed a sham operation on 10 rats. In addition, the BDL rats received 0.1 mL of vitamin K (100 μg/mL) every 4 d by subcutaneous injection. Body weight was measured daily. The animals were sacrificed in random order between 9:00 am and 11:00 am after an overnight fast. Urine samples were collected from each rat during overnight fasting using a metabolic cage. Serum, bile (from the bile cyst in the BDL animals), and liver samples were collected for further analyses as previously described16.
Plasma biochemistry and liver histology
Plasma alanine transaminase (ALT), aspartate transaminase (AST), total cholesterol (CHO), high-density lipoprotein (HDL) cholesterol and low-density lipoprotein (LDL) cholesterol, total bile acid (TBA), total bilirubin, bile and urine were analyzed using a Hitachi 7170 chemistry analyzer with kits obtained from Zhongsheng Beikong Biotechnology (Beijing, China). Formalin-fixed tissue was paraffin-embedded, and sections were stained with hematoxylin and eosin and Sirius Red. Liver necrosis and fibrosis were quantified in a blinded fashion using a Leica DM1000 microscope. For each animal, 10 images were taken at low magnification (10×objective lens) and used for quantifying the percentage of fibrotic tissue using Image J Software (NIH open source; http://rsbweb.nih.gov/ij/) with thresholding. Bile duct proliferation and liver inflammation were blindly assessed on a 1 to 4+ scale. The liver hydroxyproline concentration was measured as previously described17.
Cell culture and treatment
A human hepatic stellate cell line, LX-2, was a gift from Dr Scott Friedman (Mount Sinai Medical Center, New York, NY, USA). Cells were maintained in DMEM/GlutaMAX I supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere containing 5% CO2. When the cells reached 50%–70% confluence, they were starved in serum-free DMEM/GlutaMAX I for 24 h prior to treatment with TGF-β1, LY294002 (dissolved in DMSO) and/or EGCG (dissolved in water) in serum-free DMEM.
Real-time PCR
Real-time PCR master mix was purchased from Roche (Indianapolis, IN, USA). ABI TaqMan primers/probes were purchased from Applied Biosystems (Foster City, CA, USA). Total RNA was extracted from cells using TRIzol (Invitrogen, Carlsbad, CA, USA) and purified using a NucleoSpin RNA Clean-up Kit (Macherey-Nagel, Duren, Germany). Complementary DNA was generated using an AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies, Santa Clara, CA, USA), and the relative expression levels of specific genes were determined with an ABI 7500 Fast Real-Time PCR System. The GAPDH gene was used as a reference to normalize the data.
Western blotting analysis
Cells or homogenized tissues were lysed in RIPA buffer (50 mmol/L Tris-HCl, pH 7.5, 1% NP-40, 0 mmol/L NaCl, 1 mg/mL aprotinin, 1 mg/mL leupeptin, 1 mmol/L Na3VO4, and 1 mmol/L NaF) at 4 °C for 30 min. Cell debris was removed by centrifugation at 12000×g for 20 min at 4 °C. Protein concentrations were determined by the Bradford assay. Equal amounts of lysate were resolved by SDS-polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk in TBS-T buffer at room temperature for 1 h and then incubated for 2 h or overnight with primary antibodies. The membranes were then incubated for 1 h with an appropriate horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology). Electrochemiluminescence was performed according to the manufacturer's instructions with a ChemiImager 5500 imaging system (Alpha Innotech Corporation, San Leandro, CA, USA).
G3BP1 siRNA and plasmid transfection
siRNAs were synthesized by Ribobio Technology (Guangzhou, China). The G3BP1-siRNA targeting sequence was 5′-GCAACAGUAUUUCGGUAUAdTdT-3′. The full-length cDNA sequence of G3BP1 was constructed in a pCDNA3.1 plasmid vector. Cells were transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) or with plasmids using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions.
Statistical analysis
The data are expressed as the mean±standard deviation (SD). Differences between experimental groups were assessed for significance using two-tailed Student's t-test. A P value of less than 0.05 was considered to be statistically significant.
Results
EGCG improved the growth rate and liver histology in BDL rats
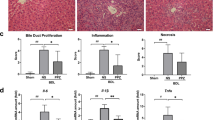
To determine whether EGCG has any beneficial effects on cholestatic liver injury, we administered EGCG to BDL rats. As shown in Figure 1A, EGCG essentially normalized the growth rate of the BDL rats compared with sham treated controls. In contrast, the growth rate of the saline-treated BDL rats was significantly lower (86.4% of the sham control group). Serum biochemistry analysis did not detect significant differences in the levels of ALT, AST, or bile acid between the EGCG- and saline-treated groups. However, a blinded assessment of liver histology revealed that the EGCG treatment significantly reduced necrosis and inflammation compared with the saline-treated rats (Figure 1B, Table 1), whereas there was no difference in bile duct proliferation between the EGCG-treated group and saline-treated BDL control group (data not shown). The reduced liver fibrosis was confirmed by Sirius Red staining and measurement of the hydroxyproline concentration (Figure 1C, 1D). These observations suggested that EGCG had hepatoprotective and anti-fibrotic effects on cholestatic liver injury.

EGCG markedly improved the growth rate and liver functioning in bile duct-ligated (BDL) rats. (A) Changes in body weight. (B) Representative H&E liver histology (40×) and blinded quantitative assessment of liver necrosis and inflammation. (C) Sirius Red staining and (D) liver hydroxyproline analysis demonstrated reduced liver fibrosis in the livers of EGCG-treated BDL rats. bP<0.05 vs the sham-operated group. eP<0.05 vs the BDL group. n=10 in each group.
EGCG significantly repressed liver fibrogenic gene expression in BDL rats
To assess gene expression associated with liver inflammation and fibrosis, we detected the hepatic mRNA and/or protein expression of TNF-α, IL-1β, TGF-β1, MMP-9, α-SMA, and COL1A1. As shown in Figure 2A, the EGCG treatment significantly reduced the mRNA expression of TNF-α, TGF-β1, and MMP-9 compared with the saline-treated BDL rats. The expression levels of the fibrotic markers α-SMA and desmin were decreased in the EGCG-treated rat livers as determined by immunohistochemistry (Figure 2B). Western blotting also detected lower levels of protein expression of α-SMA, collagen I, and TGF-β1/2 in the EGCG-treated BDL livers (Figure 2C). These findings indicated that the EGCG treatment greatly reduced fibrosis formation in this rat model of cholestasis.

EGCG significantly reduced the expression of genes involved in liver fibrosis and inflammation in BDL rat livers. (A) mRNA expression of COL1A1, MMP-2, MMP-9, TNF-α, IL-1β, and TGF-β1 in the sham, BDL, and EGCG-treated BDL livers. The mRNA expression levels were normalized against GAPDH. (B) Representative images of liver tissue sections labeled with an anti-desmin antibody or with an anti-α-SMA antibody. The positive areas were quantified using Image J Software. (C) Western blotting analysis of α-SMA, collagen 1a1 (COL1a1) and TGF-β1 expression in BDL rat livers, and the quantification of the whole group. bP<0.05 vs the sham group. eP<0.05 vs the BDL group. n=10 in each group
EGCG reduced liver fibrogenic gene expression in human hepatic stellate LX-2 cells
To determine whether EGCG has any anti-fibrotic effects on human hepatic stellate cells, we treated LX-2 cells from a well characterized and widely used human stellate cell line established by Xu18 with EGCG. As demonstrated in Figure 3A and 3B, EGCG repressed COL1A1 mRNA and protein expression levels in a dose-dependent manner when LX-2 cells were stimulated with TGF-β1. At a concentration of 25 μmol/L, EGCG reduced the COL1A1, MMP-2, TIMP-1, and TGF-β1 mRNA levels by approximately 80%. Decreased protein expression levels were also detected for TGF-β1, α-SMA, MMP-9, and MMP-2 in these cells. These results indicate that EGCG is also a strong liver anti-fibrogenic agent for human hepatic stellate cells.

EGCG reduced the expression of liver fibrogenic genes and the activity of the TGF-β/Smad signaling pathway in a dose-dependent manner in human hepatic stellate LX-2 cells. (A) mRNA expression of COL1A1, MMP-2, TIMP-1, TGF-β1, TGFBR2, and SMAD4 in LX-2 cells. bP<0.05 vs the TGF-β1 treatment group. (B) Western blotting detected the protein expression of COL1A1, TGF-β1/2, SMA, MMP-2, and MMP-9 in LX-2 cells. β-actin served as a loading control. (C) Western blotting analysis of protein expression of members of the TGF-β1/Smad signaling pathway and the protein expression of nuclear factor κB (NF-κB). β-Actin served as a loading control.
Altered G3BP1 expression did not change the anti-fibrogenic activity of EGCG in LX-2 cells.
A previous report has indicated that G3BP1 is a direct target of EGCG in lung cancer repression15. To determine whether the anti-fibrogenic effect of EGCG in LX-2 cells is mediated through G3BP1, we manipulated G3BP1 expression in these cells. First, we detected its expression. As shown in Figure 4A, TGF-β1 markedly stimulated G3BP1 protein expression in the LX-2 cells, and the addition of EGCG substantially repressed this stimulation. To gain further insight into the role of G3BP1 in the antifibrotic effects of EGCG, we altered G3BP1 expression in LX-2 cells by siRNA knockdown or by over-expression using a plasmid construct19. As shown in Figure 4B and 4C, neither the reduction nor over-expression of G3BP1 altered the EGCG-mediated repression of the mRNA expression of COL1A1, MMP-2, SMAD4, and TGF-β1 after TGF-β1 stimulation. These results suggested that G3BP1 was not involved in fibrogenesis and that it did not play a role in the anti-fibrotic activity of EGCG in the LX-2 cells.

G3BP expression did not alter the EGCG-mediated repression of liver fibrogenic genes in LX-2 cells. (A) EGCG repressed the protein expression of G3BP1. β-actin served as a loading control. Knockdown (B) or over-expression (C) of G3BP1 did not change the mRNA expression of liver fibrosis-related genes.
EGCG repressed TGF-β1-stimulated activation of Smad2/3 and Akt in LX-2 cells
The TGF-β1/Smad pathway is one of the key signaling pathways in liver fibrogenesis. Previous studies have also indicated that the PI3K/Akt signaling pathway plays an important role in this process. To determine the relationship between the two pathways and the anti-fibrotic effect of EGCG, we detected the protein expression and/or phosphorylation of several genes involved in these two pathways. In this experiment, EGCG not only repressed the phosphorylation of Smad2, Smad3, and Akt in LX-2 cells after TGF-β1 stimulation but also the protein expression of Smad4 and NF-κB (Figure 3C) and mRNA expression of TGFBR2 and Smad4 (Figure 3A). These results indicated that EGCG exerted its anti-fibrotic effect through repressing activation of the TGF-β1/Smad signal pathway in the hepatic stellate cells and that PI3K/Akt was involved in this activity.
LY294002 is a specific inhibitor of PI3K. To determine whether EGCG and LY294002 have any additive effects on the repression of fibrogenic gene expression in TGF-β1-stimulated LX-2 cells, we treated these cells with LY294002 alone or in combination with EGCG. As expected, LY294002 not only reduced the level of p-Akt but also those of p-Smad2, p-Smad3, α-SMA, and TGF-β1 (Figure 5B, lane 2, 3). However, when these cells were treated with both LY294002 and EGCG, no additive effects were observed. The repressive effects of LY294002 on COL1A1 and TGF-β1 were confirmed with an mRNA assay (Figure 5A). These results suggested that EGCG blocked the PI3K/Akt signaling pathway similar to LY294002 in the cells and that the phosphorylation of Smad2 and Smad3 was significantly decreased, followed by the blocking of the PI3K/Akt signaling pathway. These results suggested that the anti-fibrotic effects of EGCG occurred via the PI3K/Akt/Smad pathway. The protein level of Smad4 was down-regulated by EGCG but not by LY294002 (Figure 5B), indicating that EGCG has an additional role in repressing liver fibrogenesis.

Effects of EGCG on the PI3K/Akt/Smad signaling pathway in LX-2 cells and BDL rat livers. (A) LX-2 cells were treated with EGCG, LY294002 or both for 24 h, the mRNA expression of COL1A1 and TGF-β1 were detected by real-time PCR, and (B) the expression of liver fibrogenic protein and members of the PI3K/Akt/Smad signaling pathway were detected by Western blotting. (C) The protein expression of members of the PI3K/Akt and TGF-β/Smad signaling pathways in BDL rat livers, and the quantification of the whole group (n=10). bP<0.05 vs the sham group. eP<0.05 vs the BDL group.
Phosphorylation of Smad2, Smad3, and Akt were also reduced in EGCG-treated BDL rat livers.
Finally, to verify whether the anti-fibrotic effects of EGCG occur through the PI3K/Akt/Smad pathway, we assessed the phosphorylation of Smad2, Smad3, and Akt in rat liver tissues. Western blotting assay showed that p-Akt, p-Smad2, p-Smad3, Smad4, and NF-κB were up-regulated in the BDL livers compared with the sham livers. However, the phosphorylation of these proteins was significantly reduced in the EGCG-treated BDL livers (Figure 5C), confirming that EGCG repressed the phosphorylation of Smad2, Smad3, and Akt in vivo in the rat liver. Taken together, these results suggested that the inhibitory effect of EGCG on liver fibrogenic gene expression occurred through the repression of the PI3K/Akt/Smad pathway.
Discussion
In this study, we assessed the therapeutic effect of EGCG on liver fibrosis in vivo in the rat BDL model of cholestasis and in vitro in a human hepatic stellate cell line, LX-2. The administration of EGCG (25 mg/kg) to BDL rats by gavage resulted in (1) improved animal growth; (2) significantly reduced liver necrosis, inflammation and fibrosis; and (3) repressed mRNA and/or protein expression of liver fibrogenic genes. We also demonstrated that EGCG repressed TGF-β-induced collagen 1A1 expression in LX-2 cells. Further mechanistic experiments suggested that EGCG repressed the TGF-β1-stimulated phosphorylation of Smad2/3, which was followed by a decrease in the phosphorylation of Akt both in vitro in LX-2 cells and in vivo in BDL rat livers Therefore, we conclude that EGCG exerts anti-fibrotic effects in cholestatic disorders and propose that EGCG is a potential therapy for patients with liver fibrosis caused by cholestatic disorders.
During cholestasis, impaired bile secretion results in the accumulation of bile salts and toxins in hepatocytes and serum20. Elevated levels of these toxins cause oxidative stress and inflammation in the liver and stimulate the expression of proinflammatory cytokines and chemokines. In response to these stimuli, hepatic stellate cells are activated and initiate fibrogenesis, and the TGF-β/Smad signaling pathway plays a very important role in this process2. Therefore, it is conceivable that interference with TGF-β/Smad signaling would reduce fibrogenesis and improve liver functioning. EGCG has been reported to be a potent anti-oxidant and to have anti-inflammatory effects in vitro in cells and in vivo in animal models of several diseases. However, it has also been suggested that EGCG exerts its anti-oxidative and inflammatory effects through markedly different signaling mechanisms in different disease models and/or cell types. While G3BP1 appears to be the direct target in lung cancer cells19, our results indicate that this is not the case in human LX-2 cells because the knockdown or overexpression of G3BP1 did not alter the EGCG-mediated repression of COL1A1, TGF-β1, or MMP-2 expression (Figure 4). Instead, our findings indicate that EGCG exerts its anti-fibrotic and inflammatory effects through the PI3K/Akt signaling pathway in both LX-2 cells and the rat liver (Figure 1, 2, 3, 5), in agreement with the findings of Xiao et al9 Most importantly, the lack of an additive effect of EGCG and LY294002 (a specific inhibitor of PI3K) on the repression of the TGF-β1-stimulated phosphorylation of Smad2 and Smad3 and the expression of TGF-β1/2 in LX-2 cells suggests that down-regulation of the PI3K/Akt/Smad signaling pathway may be a direct or indirect target of EGCG in LX-2 cells. Nevertheless, verification of the EGCG-mediated repression of TGF-β1-stimulated COL1A1 expression in LX-2 cells suggests that the findings involving the BDL rats in the current study and those involving CCl4-treated rats7 and high-fat-diet-fed rats9 from previous reports have clinical implications in human liver fibrosis.
Despite the improved liver functioning in the EGCG-treated BDL rats (Figure 1), their bile acid and total cholesterol levels were not different from these in the BDL control group (Table 1). There was no difference in liver CYP7A1 mRNA expression (data not shown). These results suggest that the beneficial effects of the EGCG treatment are not mediated through bile acid metabolism. This finding differs from those of our previous study, in which the combined treatment of retinoic acid and ursodeoxycholic acid has been observed to greatly improve liver functioning in BDL rats16. However, decreased necrosis of the EGCG-treated liver indicates that EGCG may protect hepatocytes by reducing oxidative stress and inflammation because lower mRNA and/or protein expression levels of TNF-α, TGF-β, and NF-κB were detected in the BDL livers (Figure 5C).
In summary, we demonstrated that treatment with EGCG improved liver functioning in vivo in BDL rats and in vitro in a human hepatic stellate cell line, LX-2. We also revealed that the antifibrotic effects of EGCG occurred through the inhibition of the PI3K/Akt/Smad signaling pathway in both human LX-2 cells and BDL rat livers. These findings suggest that EGCG could be a new therapy for liver fibrosis in patients with chronic cholestatic disorders.
Author contribution
Dong-ke YU, Hong-wei HE, Rong-guang SHAO, and Shi-ying CAI designed the research project; Dong-ke YU, Hong-wei HE, Shuang-shuang ZHAO, Cai-xia ZHANG, Sheng-hua ZHANG, and Hao ZHANG performed the experiment; Dong-ke YU and Hong-wei HE analyzed the data; Hong-wei HE and Rong-guang SHAO contributed the reagents and materials; and Dong-ke YU, Hong-wei HE, and Shi-ying CAI wrote the manuscript.
References
Friedman SL . Hepatic fibrosis — overview. Toxicology 2008; 254: 120–9.
Puche JE, Saiman Y, Friedman SL . Hepatic stellate cells and liver fibrosis. Compr Physiol 2013; 3: 1473–92.
Hirschfield GM, Heathcote EJ, Gershwin ME . Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 2010; 139: 1481–96.
Fickert P, Stieger B . Molecular pathogenesis of chronic cholestatic liver disease: Impact on novel therapeutic approaches. Mol Aspects Med 2014; 37: 1–2.
Shanafelt TD, Call TG, Zent CS, Leis JF, LaPlant B, Bowen DA, et al. Phase 2 trial of daily, oral Polyphenon E in patients with asymptomatic, Rai stage 0 to II chronic lymphocytic leukemia. Cancer 2013; 119: 363–70.
Singh BN, Shankar S, Srivastava RK . Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol 2011; 82: 1807–21.
Yasuda Y, Shimizu M, Sakai H, Iwasa J, Kubota M, Adachi S, et al. (−)-Epigallocatechin gallate prevents carbon tetrachloride-induced rat hepatic fibrosis by inhibiting the expression of the PDGFRbeta and IGF-1R. Chem Biol Interact 2009; 182: 159–64.
Tipoe GL, Leung TM, Liong EC, Lau TY, Fung ML, Nanji AA . Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology 2010; 273: 45–52.
Xiao J, Ho CT, Liong EC, Nanji AA, Leung TM, Lau TY, et al. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur J Nutr 2014; 53: 187–99.
Sakata R, Ueno T, Nakamura T, Sakamoto M, Torimura T, Sata M . Green tea polyphenol epigallocatechin-3-gallate inhibits platelet-derived growth factor-induced proliferation of human hepatic stellate cell line LI90. J Hepatol 2004; 40: 52–9.
Higashi N, Kohjima M, Fukushima M, Ohta S, Kotoh K, Enjoji M, et al. Epigallocatechin-3-gallate, a green-tea polyphenol, suppresses Rho signaling in TWNT-4 human hepatic stellate cells. J Lab Clin Med 2005; 145: 316–22.
Nakamuta M, Higashi N, Kohjima M, Fukushima M, Ohta S, Kotoh K, et al. Epigallocatechin-3-gallate, a polyphenol component of green tea, suppresses both collagen production and collagenase activity in hepatic stellate cells. Int J Mol Med 2005; 16: 677–81.
Yumei F, Zhou Y, Zheng S, Chen A . The antifibrogenic effect of (−)-epigallocatechin gallate results from the induction of de novo synthesis of glutathione in passaged rat hepatic stellate cells. Lab Invest 2006; 86: 697–709.
Zhen MC, Huang XH, Wang Q, Sun K, Liu YJ, Li W, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses rat hepatic stellate cell invasion by inhibition of MMP-2 expression and its activation. Acta Pharmacol Sin 2006; 27: 1600–7.
Shim JH, Su ZY, Chae JI, Kim DJ, Zhu F, Ma WY, et al. Epigallocatechin gallate suppresses lung cancer cell growth through Ras-GTPase-activating protein SH3 domain-binding protein 1. Cancer Prev Res (Phila) 2010; 3: 670–9.
He H, Mennone A, Boyer JL, Cai SY . Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology 2011; 53: 548–57.
Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol 2006; 168: 410–22.
Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005; 54: 142–51.
Zhang H, Zhang S, He H, Zhao W, Chen J, Shao RG . GAP161 targets and downregulates G3BP to suppress cell growth and potentiate cisplaitin-mediated cytotoxicity to colon carcinoma HCT116 cells. Cancer Sci 2012; 103: 1848–56.
Maillette de Buy Wenniger L, Beuers U . Bile salts and cholestasis. Dig Liver Dis 2010; 42: 409–18.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81170409, 81101720), the National S&T Major Special Project on Major New Drug Innovation (2012ZX09301002-001), the Wang Bao En Liver Fibrosis Foundation (20110026), and the Sichuan Provincial People's Hospital Doctor's Foundation (30305030828).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yu, Dk., Zhang, Cx., Zhao, Ss. et al. The anti-fibrotic effects of epigallocatechin-3-gallate in bile duct-ligated cholestatic rats and human hepatic stellate LX-2 cells are mediated by the PI3K/Akt/Smad pathway. Acta Pharmacol Sin 36, 473–482 (2015). https://doi.org/10.1038/aps.2014.155
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.155
Keywords
This article is cited by
-
Geranylgeranylacetone attenuates fibrogenic activity and induces apoptosis in cultured human hepatic stellate cells and reduces liver fibrosis in carbon tetrachloride-treated mice
BMC Gastroenterology (2018)
-
D-chiro-inositol effectively attenuates cholestasis in bile duct ligated rats by improving bile acid secretion and attenuating oxidative stress
Acta Pharmacologica Sinica (2018)
-
Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways
Scientific Reports (2017)
-
Unconjugated bilirubin elevation impairs the function and expression of breast cancer resistance protein (BCRP) at the blood-brain barrier in bile duct-ligated rats
Acta Pharmacologica Sinica (2016)
-
A comprehensive overview of hepatoprotective natural compounds: mechanism of action and clinical perspectives
Archives of Toxicology (2016)
