
Abstract
Aim:
To determine the postshock activation patterns with both successful and failed shocks in a canine model of ventricular fibrillation, and whether piniacidil, an early after-depolarization (EAD) inhibitor, altered the defibrillation threshold (DFT) and postshock activation patterns.
Methods:
In 6 beagles, a basket catheter with 64 unipolar electrodes was placed in the LV for global endocardial mapping, a monophasic action potential catheter was inserted into the LV apex, and a catheter with the negative electrode in the right ventricle and the positive electrode in the superior vena cava was inserted for defibrillation. The DFT, 90% action potential duration (APD90) and activation recovery interval (ARI) were evaluated before and after pinacidil administration (loading dosage 0.5 mg/kg and maintenance dosage 0.5 mg·kg-1·h-1, iv). Electrical heterogeneities were defined with the dispersion of ARI. After successful and failed shocks with near-DFT strength, the earliest postshock activation patterns (focal or nonfocal endocardial activation), interval and location were detected.
Results:
Pinacidil significantly decreased APD90 (from 178±16 ms to 168±18 ms) and ARI from (152±10 ms to 143±10 ms) at pacing cycle length of 300 ms. The drug significantly increased VF activation rate (from 10.0±1.9 Hz to 10.8±2.0 Hz). The drug did not affect the dispersion of ARI, neither it changed DFT (baseline: 480±110 V; pinacidil: 425±55 V, P>0.05). The earliest postshock activation arose locally on the LV apical endocardium before and after the drug treatment. Pinacidil significantly prolonged the postshock cycle length of cycles 2 to 5 for the successful episodes but not for the failed episodes.
Conclusion:
Pinacidil increases the postshock cycle length suggesting that EAD may play a role in postshock activation, while it fails to alter DFT suggesting that EAD produced by shock does not determine a defibrillation success or failure.
Similar content being viewed by others
Introduction
Although electrical shock is the most effective strategy for terminating ventricular fibrillation (VF), the mechanism of defibrillation is not yet fully understood1,2. Reentry has been shown to be responsible for defibrillation failure of low strength shocks3. With the shock strength increasing to near defibrillation threshold (DFT), focal activation is responsible for failed defibrillation4,5,6,7. The earliest postshock focal activations arise from the lowest shock gradient area8. For a shock delivered from right ventricle to superior vena cava, the lowest shock gradient area is located at the left ventricular free wall8, this is also where the earliest postshock activations arise. The earliest postshock focal activations might be caused by microreentry9, triggered activity10, or tunnel propagation6,8. For a strong electrical shock to reinitiate reentry, the shock should create a uni-directional block and slow conduction area, an area that usually forms near the location of the shock electrodes11,12. Thus, reentry is less likely to be the main mechanism for a postshock earliest activation that arises far away from the shock electrodes. The triggered activity has been proposed as a mechanism responsible for the earliest postshock activation4. Delayed after-depolarization (DAD) mediated triggered activities in Purkinje fibers have been shown to cause recurrence of ventricular arrhythmias following successful defibrillation with the shock electrodes located at left ventricle (LV) and right ventricle (RV)10. While epicardial electrical mapping study in pig did not find evidence that activation following failed defibrillation caused by triggered activity5, it is not clear whether triggered activity from the endocardium plays a role in postshock activation, because the Purkinje system is distributed endocardially in the human and canine hearts. Thus, the purpose of this study was to determine the postshock activation patterns with both successful and failed shocks and how piniacidil, an early after-depolarization (EAD) inhibitor, altered the DFT and the postshock activation patterns in canine model.
Materials and methods
All animals were purchased from Shanghai Jiao Tong University Agriculture College of China and raised under controlled conditions in the Department of Animals for Scientific Research, Shanghai Jiao Tong University School of Medicine. All procedures were approved by the Animal Protection Committee of Shanghai Jiao Tong University.
Animals
Six beagles (11±1.2 kg, mean±SD) were injected intramuscularly with ketamine (10 mg/kg) and atropine (0.04 mg/kg) for anesthetic induction. Anesthetization was maintained with propofol (8–12 mL·kg-1·h-1) and the animal was ventilated in a restrained, dorsally recumbent position. Systolic blood pressure (SBP), arterial blood gases, ECG, and serum electrolytes were monitored throughout the entire study. The heart was exposed through a median sternotomy and supported in a pericardial sling.
Electrode placement
A catheter (model 80993, IBI, ST JUDE MEDICAL) with the negative electrode in the right ventricle and the positive electrode in the superior vena cava was inserted for defibrillation. A 31 mm multi-electrode basket (Constellation Catheter, model US8031U, Boston Scientific, Natick, MA, USA) was introduced through a carotid artery into the LV (Figure 1A). The catheter contained eight splines each with eight electrodes approximately 2 mm apart. Figure 1-II shows the basket orientation and the division of the basket electrodes into regions for statistical analyses. A monophasic action potential (MAP) catheter (Boston Scientific, San Jose, CA, USA) was positioned through a femoral artery in the LV.

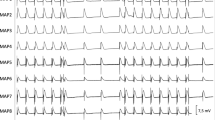
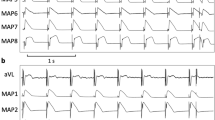
Global mapping of left ventricular endocardium. (A) Fluoroscopic image of a posterior-anterior view of the LV basket catheter, the RV defibrillation catheter and MAP catheter. (a) MAP catheter; (b) basket catheter; (d)defibrillation catheter. (B) Display of the basket orientation in the LV: (I) anterior free wall; (II) lateral free wall; (III) posterior free wall; (IV)septum. Apical electrodes are toward the center of the display (a), and basal electrodes are toward the periphery (h). For statistical analysis, first activation locations during the earliest postshock activation cycle after shocks were grouped into regions i–viii (i–iv, basal region; v-viii, apical region). (C) shows the tracings of lead (B) of ECG, one of the basket electrodes and APD recorded at the pacing cycle length of 300 ms. A solid vertical line marks the onset of stimulus artifact.
Defibrillation protocol
VF was induced by a 30-Hz stimulation delivered (MicroPace III, EPS 320 Cardiac stimulator) through the pacing electrode in the RV. After 20 s of electrically induced VF, a biphasic shock (6/4 ms) was delivered from a defibrillator (Teletronic Pacing Systems, 4510 Implant Support Device) via the defibrillating catheter electrodes. The leading edge voltage of the first shock was 400 V for the first animal. Depending on the success or failure of the shock, the leading edge voltage was decreased or increased by 40 V, respectively. The transition from failure to success or success to failure was recorded as the first data point. The up-down algorithm was continued until the third reversal of success to failure or failure to success was reached. The DFT was determined by averaging the four shock strengths that formed the three reversals. A failed shock with a 10% lower voltage than the DFT and a successful shock with 10% stronger than the DFT were delivered respectively following the DFT determination. VF recurrence was defined as VF occurring more than 5 s after successful defibrillation of VF. At least 5 min elapsed after every VF episode to allow blood pressure and heart rate to return to normal.
Action potential duration and activation recovery interval measurements
To determine the action potential duration (APD), the LV apex was stimulated with a series of 30 stimuli of 300 ms through the tip of the basket catheter. Pacing stimuli were delivered at twice diastolic threshold. The APD at 90% repolarization (APD90) was measured from the last of the 30 action potentials recorded by the MAP electrode in the LV (Figure 1C). The activation recovery interval (ARI) was determined to estimate the APD and, hence, the refractory period13. Activation time in the unipolar electrograms was defined as the steepest downslope of the QRS complex and recovery time as the fastest upslope of the T wave. The ARI was defined as the interval between the activation time and the recovery time. The unipolar recordings were discarded from measurement because of electrograms in which the fastest upslope of the T wave was ambiguous. APD90 and ARI were determined before and after administration of pinacidil.
The QT interval of ECG was determined in sinus rhythm and at the pacing cycle length (PCL) of 300 ms before and after pinacidil.
VF activation rate and VF cycle length
The activation rate was estimated by Fast Fourier Transform (FFT) analysis of VF at each electrode of the basket catheter. The frequency with the highest power between 2 and 20 Hz was taken as the activation rate. The VF cycle length (VF-CL) was defined as the reciprocal of the activation rate.
Definition of postshock interval and focal activation pattern
Earliest activation was defined as the earliest activation front recorded after the shock that propagated throughout the myocardium, and the electrode that first recorded this activation front was labeled the earliest activation site5. The postshock interval was defined as the interval from the beginning of the shock until the first postshock activation at the earliest activation site. The four successive cycles following the first postshock activation also were analyzed.
Focal activation was defined as an activation sequence that spread in all directions centrifugally away from its site of earliest activation5. Nonfocal activation was defined as an activation sequence that spread in some directions but not others away from its site of earliest activation5.
Spatial heterogeneity
ARI dispersions were quantified using the coefficient of variation (CoV, the standard deviation divided by the mean) from all the unipolar electrodes.
Pinacidil administration
After the baseline parameters were measured, pinacidil (loading dosage 0.5 mg/kg in 10 min, maintenance dosage 0.5 mg·kg-1·h-1) was administered intravenously in six animals. Thirty minutes after giving the loading dose, the parameters were again determined. The infusion of pinacidil was maintained until the end of the experiment.
Statistical analysis
Results were expressed as mean±SD. VF activation rate, VF-CL, postshock intervals, blood pressure, QT interval, DFT, APD90, and ARI before and after drug treatment were compared by paired Student's t-test. The earliest postshock activation location frequency was compared for electrodes closer to LV apex (v–viii, Figure 1B) and for electrodes closer to LV base (i–iv) with a X2 distribution. For all analyses P<0.05 was considered statistically significant.
Results
Pinacidil significantly decreased SBP by 41% (Table 1) at 30 min after the loading dose was given (Table 1). Pinacidil significantly abbreviated the QT interval at pacing and sinus rhythm compared to the baseline (P<0.05, Table 1). APD90 and ARI were also significantly shortened by pinacidil at pacing and sinus rhythm (P<0.01, Table 1). Pinacidil significantly increased the VF activation rate (P<0.01, Table 1). These results suggested the drug had its expected electrophysiological effects.
Compared to baseline, the pinacidil did not significantly change the spatial dispersion of refractoriness (P>0.05, Table 1, Figure 2). The delivered leading-edge voltage and energy of DFT at baseline was 480±110 V and 14±3.2 J, respectively (Table 1). After the pinacidil was given, the delivered leading-edge voltage and energy of DFT were 425±55 V and 11.5±1.5 J, respectively. There were no statistical differences for voltage and energy DFT before or after pinacidil administration.

Representative maps of ARIs dispersion before and after pinacidil treatment. Representative maps of ARIs at the PCL of 300 ms in one animal at baseline (A) and after pinacidil (B). The ARI at each electrode is represented by a color corresponding with the value ranges shown on the bar. The ARIs have been normalized by dividing the ARI at each electrode by the mean ARI for all electrodes.
Postshock intervals of failed defibrillation shocks after pinacidil were not significantly different from those before the drug treatment (78±20 vs 88±21 ms, P>0.05). No episode of VF recurrence was observed after successful defibrillation of 20 s-VF before and after pinacidil administration in this study. Five out of six earliest activations following failed shocks, both before and after administration of pinacidil, arose from the apical area of LV endocardium (Figure 3). The incidence of the location site for the earliest activation was not significantly altered by the drug. After administration of pinacidil, in 5 of the 6 failed shock episodes earliest activation wavefronts spread in a focal pattern (Figure 4A, 4B), while in the remaining 1 episode earliest activation wavefronts spread in a nonfocal pattern. The relative incidence of focal and nonfocal patterns was not significantly changed by the administration of pinacidil.

Locations of earliest postshock activation following successful and failed defibrillation shocks. (A) shows the locations of first postshock activation cycles after failed near-DFT shocks, and (B) shows the locations of first postshock activation cycles following successful defibrillation shocks. For statistical analysis, earliest postshock activation cycles were grouped as shown in Figure 1B.

Earliest postshock activation maps of successful and failed defibrillation shocks. (A and B) Show the activation maps of the earliest postshock activation cycles following failed defibrillation shocks before and after pincaidil. (C and D) Show the activation maps of the first postshock activation cycles following successful defibrillation shocks before and after the drug. Activation times are indicated by the color bar. Triangles indicate the site of earliest activation in each panel.
Two types of postshock intervals of successful defibrillation shocks were observed: Type A successes, immediate termination as indicated by a long postshock interval (>130 ms); and Type B successes, a short postshock interval (≤130 ms) followed by one to five rapid ectopic cycles14. Both types of postshock interval were detected at baseline (Type A, 202±53 ms; Type B, 108±11 ms) and after pinacidil (Type A, 204±101 ms; Type B, 95±5 ms). The ratio of Type A and Type B successes was not significantly changed by pinacidil (2/6 vs 3/6, P>0.05). Similar to the failed defibrillation shocks, most of (5/6) the earliest activation locations following successful defibrillation shocks were on the LV apical endocardium (Figure 3) and propagated toward the base both at baseline and after pinacidil treatment. Successful shock episodes earliest activation always appeared focally before and after administration of pinacidil (Figure 4III, 4IV).
The postshock cycle length of cycles 2 to 5 were shorter in failed episodes than in successful episodes (P<0.01, Figure 5) for both baseline and pinacidil indicating that successive postshock cycles arose more rapidly in failed than successful episodes. The drug significantly prolonged postshock cycle length of cycles 2 to 5 for successful episodes but not for failed episodes (Figure 5).

Postshock interval and inter-cycle interval after successful and failed defibrillation shocks. (A) Shows five successive postshock interval with the standard deviation indicated by an error bar after successful and failed defibrillation. The postshock cycle length of cycles 2 to 5 were shorter in failed episodes than in successful episodes for both baseline and pinacidil. (B) The drug significantly prolonged postshock inter-cycle interval of cycles 2 to 5 for successful episodes but not for failed episodes.
Discussion
The main findings of this study are as follows: (1) Pinacidil significantly prolonged postshock cycle length of cycles 2 to 5 for successful episodes but not for failed episodes.; (2) Pinacidil, an EAD inhibitor, did not significantly change the location or time of earliest postshock activation or the patterns of activation spread away from the early site; (3) Consistent with a previous study5 pinacidil did not significantly alter the DFT. These results indicate EAD may play a role in postshock activation. However, the EAD produced by shock does not determine a defibrillation success or failure.
Alteration of APD and QT interval by pinacidil
The KATP channel is activated by a reduction in ATP that occurs during acute ischemia and has been implicated in early ischemic changes in electrophysiological parameters15. The increase in outward potassium conductance results in the shortening of repolarization and APD16,17,18. Pinacidil, an ATP-sensitive potassium channel opener, has effects similar to those of ischemia on action potentials. In this study, the drug significantly shortened the APD and ARI as expected (Table 1). The shortened refractory period probably led to an increase of activation rate and a decrease of VF-CL which was observed after administering pinacidil (Table 1). These findings suggested that the drug had its expected electrophysiological effects.
Pinacidil has been documented to decrease19,20 or cause no change18 in the QT interval of ECG in animal studies. Generally, the QT interval is determined by APD of ventricular myocytes21. Padrini et al described, pinacidil (10–50 μmol/L) shortened the QT interval by 10%–30% in isolated pig hearts19. However, another study reported that pinacidil significantly shortened APD but not the QT interval in isolated rabbit hearts18. Pinacidil doesn't have significant direct effects on Ca2+channels, and the QT interval on extracellular electrograms maintains a close relationship with the duration of Ca2+ transient, thus, pinacidil shortened APD to a much greater extent than it shortened the QT interval. In our study, pinacidil significantly shortened the QT interval in in vivo canine hearts. The mechanism responsible for the different QT interval effects of pinacidil in different studies are not well known but may be related to the different species, experimental models and dosages of pinacidil administration.
The earliest postshock activation patterns in our study
Consistent with previous studies22,23,24, the site of earliest postshock activation near-DFT strength delivered from electrodes placed in the RV apex and the superior vena cava is at the LV apex, which is the region exposed to the weakest potential gradient for this lead configuration. Due to advanced optical and computer simulation techniques, it is well understood that a defibrillation shock simultaneously depolarizes and hyperpolarizes cardiac tissue, known as the virtual electrode polarization25. The virtual electrode creates graded responses that lead to new activation and wavefront propagation26. A special pathway (the tunnel propagation) should exist to allow the wavefront to propagate to the low gradient region after the shock6. Earliest postshock activation in canine was within the endocardial half of the LV wall in 79% of the shock episodes, but was in the epicardial half in 84% of episodes in swine27. This different distribution of earliest postshock activation in the two species draws our attention to the Purkinje fibers involved in the creation and propagation of earliest activation, because in the pig the Purkinje fibers extend from the endocardium almost to the epicardium, while in dogs the Purkinje fibers are limited to the subendocardium. Dosdall et al28 demonstrated that the Purkinje system is active during the early postshock activation cycles following DFT level shocks with the same shock electrode configuration as ours. Therefore, the endocardial basket in the LV is capable of detecting the earliest activation pattern after the defibrillation shock in the canine model.
Maruyama et al10 recently recorded the DAD in the Purkinje system to initiate postshock arrhythmia in Langendorff-perfused rabbit ventricles. Although the findings are not directly applicable to our study because they used LV and RV electrodes to deliver the defibrillation shock and the arrhythmia arose immediately after the shock, thus, the mechanism of postshock arrhythmia may not be the same as arrhythmias that arise from the weakest potential gradient region after an isoelectric window. These findings indicate that vulnerability to triggered activity is higher in Purkinje fibers than ventricular myocardium in intact whole hearts. The majority of postshock activations appear to originate from a focal source or a breakthrough on the epicardial surface after a postshock quiescent period29,30,31,32. If the triggered activity is responsible for the earliest activation and induction of VF, then the long quiescent period after a defibrillation shock facilitates the occurrence of EADs as opposed to DADs. The increased postshock cycle length of cycles 2 to 5 following successful defibrillation by pinacidil supports our hypothesis. Pinacidil might successfully inhibit the EAD to trigger new activations. Pinacidil did not prolong the 5 postshock activation intervals of the failed defibrillation might indicate the EADs have not been inhibited by pinacidil in those cases. One potential explanation for this observation is the EAD is only one of many mechanisms for postshock activation arrhythmias. Another potential explanation is that the dosage was not sufficient to inhibit EAD mediated triggered activity. While, pinacidil had exerted the expected electrophysiological effects on the ventricular myocardium including shortened APD and ARI, the dosage for pinacidil to inhibit the EAD might be higher than its dosage to reach electrophysiological effect.
Electrophysiological mapping can identify a true focus and not confuse it with macroreentry if the spacing between electrical sensors is less than 2 mm. Although this spacing is feasible in basic experimental studies, it may not always be so during the clinical mapping of arrhythmia. In clinical practice, macroreentrant circuits tend to have a stable cycle length with little beat-to-beat variability. Small reentrant circuits are typically associated with highly fractionated electrodgrams that are usually of small amplitude. Based on those finding, our data highly suggested the triggered activities might be the main mechanism of post-shock activity.
It is well known that the heterogeneities in refractory periods that contribute to the transition from VT into VF for various cardiac disease by reentry mechanism. Treatment directed at ventricular fibrillation cycle length dispersion may be useful in the suppression of ventricular fibrillation recurrence following cardioversion. We did not find the pinacidil alter the dispersion of refractoriness in the study, hence somehow limited the role of reentry in postshock activities.
Limitations
Several limitations in this study are as follows. A limitation of this study is that we only focused on the healthy canine heart. Many pathological changes will impact the effect of anti-arrhythmic drugs on the heart. Defibrillation mechanism may differ between normal hearts and diseased hearts. Whether the same effects of pinacidil on postshock activation are associated with myocardial infarction or heart failure should be examined in future studies. Another limitation of this study is that we used only one pinacidil dosage. Although it is a dosage that has been shown to have expected effects on myocardial electrophysiologic properties, it is not well known if the dosage was sufficient to affect the Purkinje fibers.
A third limitation of this study is that we did not perform intramural recordings. It is possible that the EAD inhibitor could have altered earliest activation before it broke through to the endocardium. However, intramural recordings indicate that earliest postshock activation for shocks of near-DFT strength in dogs should be mostly recorded on the endocardium. Thus, it would be possible that the changes would also have been observed on the endocardium even if the postshock activations had been significantly altered. Also, this study did not determine if the earliest postshock activation was initiated within the specialized conduction system or propagated retrogradely into it from the working myocardium.
Author contribution
Qi JIN carried out the electrophysiological mapping studies, performed the statistical analysis and drafted the manuscript. Ning ZHANG and Jian ZHOU carried out the mapping study. Chang-jian LIN and Yang PANG performed the statistical analysis. Gang GU participated in the design of the study. Wei-feng SHEN and Li-qun WU conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
References
Dosdall DJ, Fast VG, Ideker RE . Mechanisms of defibrillation. Annu Rev Biomed Eng 2010; 12: 233–58.
Chattipakorn N, Ideker RE . The vortex at the apex of the left ventricle: a new twist to the story of the electrical induction of rotors? J Cardiovasc Electrophysiol 2003; 14: 303–5.
Kwaku KF, Dillon SM . Shock-induced depolarization of refractory myocardium prevents wave-front propagation in defibrillation. Circ Res 1996; 79: 957–73.
Chattipakorn N, Fotuhi PC, Chattipakorn SC, Ideker RE . Three-dimensional mapping of earliest activation after near-threshold ventricular defibrillation shocks. J Cardiovasc Electrophysiol 2003; 14: 65–9.
Zheng X, Walcott GP, Smith WM, Ideker RE . Evidence that activation following failed defibrillation is not caused by triggered activity. J Cardiovasc Electrophysiol 2005; 16: 1200–5.
Ashihara T, Constantino J, Trayanova NA . Tunnel propagation of postshock activations as a hypothesis for fibrillation induction and isoelectric window. Circ Res 2008; 102: 737–45.
Hwang GS, Hayashi H, Tang L, Ogawa M, Hernandez H, Tan AY, et al. Intracellular calcium and vulnerability to fibrillation and defibrillation in Langendorff-perfused rabbit ventricles. Circulation 2006; 114: 2595–603.
Constantino J, Long Y, Ashihara T, Trayanova NA . Tunnel propagation following defibrillation with ICD shocks: hidden postshock activations in the left ventricular wall underlie isoelectric window. Heart Rhythm 2010; 7: 953–61.
Ideker RE, Rogers JM, Fast V, Li L, Kay GN, Pogwizd SM . Can mapping differentiate microreentry from a focus in the ventricle? Heart Rhythm 2009; 6: 1666–9.
Maruyama M, Joung B, Tang L, Shinohara T, On YK, Han S, et al. Diastolic intracellular calcium-membrane voltage coupling gain and postshock arrhythmias: role of purkinje fibers and triggered activity. Circ Res 2010; 106: 399–408.
Qu F, Li L, Nikolski VP, Sharma V, Efimov IR . Mechanisms of superiority of ascending ramp waveforms: new insights into mechanisms of shock-induced vulnerability and defibrillation. Am J Physiol Heart Circ Physiol 2005; 289: H569–577.
Frazier DW, Wolf PD, Wharton JM, Tang AS, Smith WM, Ideker RE . Stimulus-induced critical point. Mechanism for electrical initiation of reentry in normal canine myocardium. J Clin Invest 1989; 83: 1039–52.
Jin Q, Chen X, Smith WM, Ideker RE, Huang J . Effects of procainamide and sotalol on restitution properties, dispersion of refractoriness, and ventricular fibrillation activation patterns in pigs. J Cardiovasc Electrophysiol 2008; 19: 1090–7.
Chen PS, Shibata N, Dixon EG, Wolf PD, Danieley ND, Sweeney MB, et al. Activation during ventricular defibrillation in open-chest dogs. Evidence of complete cessation and regeneration of ventricular fibrillation after unsuccessful shocks. J Clin Invest 1986; 77: 810–23.
Deutsch N, Klitzner TS, Lamp ST, Weiss JN . Activation of cardiac ATP-sensitive K+ current during hypoxia: correlation with tissue ATP levels. Am J Physiol 1991; 261: H671–676.
Wilde AA, Escande D, Schumacher CA, Thuringer D, Mestre M, Fiolet JW, et al. Potassium accumulation in the globally ischemic mammalian heart. A role for the ATP-sensitive potassium channel. Circ Res 1990; 67: 835–43.
Jones DL, Kim YH, Natale A, Klein GJ, Varin F . Bretylium decreases and verapamil increases defibrillation threshold in pigs. Pacing Clin Electrophysiol 1994; 17: 1380–90.
Wu S, Hayashi H, Lin SF, Chen PS . Action potential duration and QT interval during pinacidil infusion in isolated rabbit hearts. J Cardiovasc Electrophysiol 2005; 16: 872–8.
Padrini R, Bova S, Cargnelli G, Piovan D, Ferrari M . Effects of pinacidil on guinea-pig isolated perfused heart with particular reference to the proarrhythmic effect. Br J Pharmacol 1992; 105: 715–9.
Milberg P, Tegelkamp R, Osada N, Schimpf R, Wolpert C, Breithardt G, et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol 2007; 18: 658–64.
Yan GX, Lankipalli RS, Burke JF, Musco S, Kowey PR . Ventricular repolarization components on the electrocardiogram: cellular basis and clinical significance. J Am Coll Cardiol 2003; 42: 401–9.
Chattipakorn N, Banville I, Gray RA, Ideker RE . Mechanism of ventricular defibrillation for near-defibrillation threshold shocks: a whole-heart optical mapping study in swine. Circulation 2001; 104: 1313–9.
Zhou X, Daubert JP, Wolf PD, Smith WM, Ideker RE . Epicardial mapping of ventricular defibrillation with monophasic and biphasic shocks in dogs. Circ Res 1993; 72: 145–60.
Ashihara T, Namba T, Yao T, Ozawa T, Kawase A, Ikeda T, et al. Vortex cordis as a mechanism of postshock activation: arrhythmia induction study using a bidomain model. J Cardiovasc Electrophysiol 2003; 14: 295–302.
Trayanova NA, Gray RA, Bourn DW, Eason JC . Virtual electrode-induced positive and negative graded responses: new insights into fibrillation induction and defibrillation. J Cardiovasc Electrophysiol 2003; 14: 756–63.
Cheng Y, Mowrey KA, Van Wagoner DR, Tchou PJ, Efimov IR . Virtual electrode-induced reexcitation: A mechanism of defibrillation. Circ Res 1999; 85: 1056–66.
Fang X, Walcott GP, Huang J, Melnick S, Killingsworth CR, Smith WM, et al. The transmural location of earliest activation following a defibrillation shock is species dependent. Heart Rhythm 2005; 2: S86–S87.
Dosdall DJ, Cheng KA, Huang J, Allison JS, Allred JD, Smith WM, et al. Transmural and endocardial Purkinje activation in pigs before local myocardial activation after defibrillation shocks. Heart Rhythm 2007; 4: 758–65.
Shibata N, Chen PS, Dixon EG, Wolf PD, Danieley ND, Smith WM, et al. Influence of shock strength and timing on induction of ventricular arrhythmias in dogs. Am J Physiol 1988; 255: H891–901.
Gray RA, Jalife J . Effects of atrial defibrillation shocks on the ventricles in isolated sheep hearts. Circulation 1998; 97: 1613–22.
Idriss SF, Wolf PD, Smith WM, Ideker RE . Effect of pacing site on ventricular fibrillation initiation by shocks during the vulnerable period. Am J Physiol 1999; 277: H2065–2082.
Chattipakorn N, KenKnight BH, Rogers JM, Walker RG, Walcott GP, Rollins DL, et al. Locally propagated activation immediately after internal defibrillation. Circulation 1998; 97: 1401–10.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No 81070266 and 81000081); Shanghai Science and Technology Committee Grants (No 10140903100); Program for Innovative Research Team of Shanghai Municipal Education Commission.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jin, Q., Zhang, N., Zhou, J. et al. The effect of pinacidil on postshock activation and ventricular defibrillation threshold in canine hearts. Acta Pharmacol Sin 33, 1488–1494 (2012). https://doi.org/10.1038/aps.2012.96
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.96
