
Abstract
Aim:
To investigate the anti-inflammatory effect of Z-ligustilide (LIG) on lipopolysaccharide (LPS)-activated primary rat microglia.
Methods:
Microglia were pretreated with LIG 1 h prior to stimulation with LPS (1 μg/mL). After 24 h, cell viability was tested with MTT, nitric oxide (NO) production was assayed with Griess reagent, and the content of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and monocyte chemoattractant protein (MCP-1) was measured with ELISA. Protein expression of the nuclear factor-κB (NF-κB) p65 subunit, cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS) was detected with immunocytochemistry 1 h or 24 h after LPS treatment.
Results:
LIG showed a concentration-dependent anti-inflammatory effect in LPS-activated microglia, without causing cytotoxicity. Pretreatment with LIG at 2.5, 5, 10, and 20 μmol/L decreased LPS-induced NO production to 75.9%, 54.4%, 43.1%, and 47.6% (P<0.05 or P< 0.01), TNF-α content to 86.2%, 68.3%, 40.1%, and 39.9% (P<0.01, with the exception of 86.2% for 2.5 μmol/L LIG), IL-1β content to 31.5%, 27.7%, 0.6%, and 0% (P<0.01), and MCP-1 content to 84.4%, 50.3%, 45.1%, and 42.2% (P<0.05 or P<0.01), respectively, compared with LPS treatment alone. LIG (10 μmol/L) significantly inhibited LPS-stimulated immunoreactivity of activated NF-κB, COX-2, and iNOS (P<0.01 vs LPS group).
Conclusion:
LIG exerted a potent anti-inflammatory effect on microglia through inhibition of NF-κB pathway. The data provide direct evidence of the neuroprotective effects of LIG and the potential application of LIG for the treatment of the neuroinflammatory diseases characterized by excessive microglial activation.
Similar content being viewed by others

Introduction
Neuroinflammation characterized by the activation of glia has been closely associated with the pathogenesis of a number of neurodegenerative diseases (NDDs), including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), dementia resulting from infection with human immunodeficiency virus (HIV), and stroke1. Microglia, the resident macrophages in the normal healthy adult nervous system, form the first line of defense against injury to the central nervous system and are considered the major glia responsible for inflammation-mediated neurotoxicity. Activation of microglia is often observed in neuronal injuries and is also induced after stimulation with lipopolysaccharide (LPS), interferon (IFN)-γ, and β-amyloid in vitro2. Upon activation, microglia can induce significant and highly detrimental neurotoxic effects attributable to excess production of a wide range of proinflammatory mediators, including inflammatory enzymes (eg, inducible nitric oxide synthase [iNOS] and cyclooxygenase-2 [COX-2]), pro-inflammatory cytokines (eg, interleukin-1β [IL-1β] and tumor necrosis factor-α [TNF-α]), chemokines (eg, monocyte chemoattractant protein [MCP-1]), and transcription factors (eg, nuclear factor-κB [NF-κB])1, 2. Accumulating evidence indicates that excessive activation of microglia results in a self-perpetuating cycle of neuronal death by releasing these neurotoxic mediators1. Therefore, anti-inflammatory treatment via inhibition of microglial activation is regarded as a promising strategy for preventing NDDs and stroke in the clinic.
Z-ligustilide (3-butylidene-4,5-dihydrophthalide, LIG; Figure 1) is a major active ingredient of many medicinal plants in the Umbelliferae family3. Recent studies from our laboratory demonstrated that LIG has significant neuroprotective effects against stroke and AD through multiple mechanisms, including anti-neuroinflammation4, 5, 6, 7, 8. Whether LIG can directly regulate microglial activation through an inflammatory process has not been studied. Therefore, to further explore the anti-neuroinflammatory effects of LIG in vivo, the present study investigated the effect of LIG on the LPS-induced proinflammatory response in primary rat microglia.

Chemical structure of Z-ligustilide (LIG).
Materials and methods
Reagents
Dang gui, Angelica sinensis, was purchased from its Cultivating Base of Good Agricultural Practice in Min Xian County, Gansu Province, China. Its identity was confirmed through reference to the descriptions of the characteristics and the appropriate monograph in the Chinese Pharmacopoeia. LIG was prepared using a well-established procedure in our laboratory9. The purity was found to be >98% based on the percentage of total peak area by high-performance liquid chromatography analysis.
Bacterial LPS (Escherichia coli serotype 0111:B4) and poly-L-lysine (PLL) were purchased from Sigma-Aldrich (St Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from Amresco (Solon, OH, USA). High-glucose Dulbecco's modified Eagle's medium (DMEM) was obtained from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT, USA). 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was obtained from Sino-American Biotec (Luoyang, Henan, China). Griess reagent was obtained from Beyotime Institute of Biotechnology (Nantong, Jiangsu, China). ELISA kits were obtained from R&D Systems (Minneapolis, MN, USA). The avidin-biotin complex (ABC) kit and rabbit polyclonal anti-COX-2, anti-iNOS, and anti-NF-κB p65 antibodies were obtained from Wuhan Boster Biological Technology (Wuhan, Hubei, China). Mouse anti-rat OX-42 (CD11b) antibody was obtained from AbD Serotec (Oxford, Canterbury, UK).
LIG was dissolved in DMSO and diluted to the indicated concentrations with DMEM culture medium. Corresponding amounts of DMSO were added to the control cultures. The final DMSO concentration in control cultures (0.1%) did not show any effect on primary microglial cell cultures (data not shown). LPS was dissolved in sterile phosphate-buffered saline (PBS) and diluted with DMEM to the specified concentration.
Primary microglial culture and treatment
Primary mixed glial cultures were prepared from postnatal day 1 Sprague-Dawley rat pups as previously described10. Briefly, their cerebral cortices were aseptically excised and carefully stripped of their blood vessels and meninges in cold D-Hank's solution. Cerebral cortical fragments were dissociated by soft trituration in ice-cold DMEM with 10% FBS, 2 mmol/L glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin, resuspended in culture medium, and brought to a single cell suspension by repeated pipetting. The suspension was sequentially filtered through a 40 μm mesh, and the dissociated cells were seeded (1×106 cells per dish) on 100 mm culture dishes (Corning, Glendale, Arizona, USA) and incubated at 37 °C in humidified 5% CO2/95% air (CO2 incubator, Thermo Electron Corporation, Asheville, North Carolina, USA). The culture medium was replenished 1 day after cell plating and changed every 4–6 days. Rat microglia were obtained by shaking the primary mixed glial cultures between days 12 and 14. The floating cell suspension was collected, centrifuged at 250×g for 5 min, and resuspended in culture medium. The >96% purity of microglia (identified by OX-42-specific antibody) was used. Primary microglia were then placed into 96-well (5×104 cells/well) or 24-well (1×105 cells/well) tissue culture plates pre-coated with 0.1 mg/mL PLL and incubated at 37 °C in humidified 5% CO2/95% air. The culture medium was changed after 24 h. On the day of the experiment, the culture medium was replaced with a fresh medium containing different reagents. One hour after pretreatment with the indicated concentrations of LIG or control vehicle, the microglia were treated with LPS (1 μg/mL) or volume-matched DMEM medium.
Cellular viability assay
The effect of LIG on LPS-treated microglial viability was analyzed with the MTT assay. After microglia (5×104 cells/well) were incubated with LPS in 96-well plates for 24 h in the presence or absence of LIG at the indicated concentrations, the culture media were removed, and the cells were incubated with 5 mg/mL MTT in fresh medium at 37 °C for an additional 4 h. At the end of the incubation period, the supernatants were removed, and 150 μL DMSO was added to each well to dissolve the formed blue formazan. Cell viability was assessed by colorimetric changes using a microplate reader at 490 nm (Bio-Rad, Hercules, California, USA). Data are expressed as a percentage of LPS treatment alone.
Nitrite assays
The amount of NO secreted in cellular culture supernatants was measured by a Griess reaction. After microglia (5×104 cells/well) were stimulated with LPS in 96-well plates for 24 h, 50 μL of each culture medium was mixed with the same volume of the Griess reagent (1% sulfanilamide/0.1% N-[1-naphthyl]-ethylenediamine dihydrochloride/2.5% H3PO4), respectively, on a 96-well plate and incubated at 25 °C for 10 min. NO concentration was determined by measuring the absorbance at 570 nm on a microplate reader. Nitrite concentration was calculated with a reference to a standard curve of sodium nitrite generated by known concentrations. Data are expressed as a percentage of LPS treatment alone.
Enzyme-linked immunosorbent assay (ELISA)
Twenty-four hours after LPS stimulation, the concentrations of TNF-α, IL-1β, and MCP-1 secreted in microglia culture supernatants were measured by sandwich immunoassay according to the kit instructions. Briefly, after microglia (1×105 cells/well) were stimulated with LPS in 24-well plates for 24 h, 100 μL of each culture medium or the cytokine standard preparation were added into 96-well ELISA microplates and incubated at 37 °C, followed by washes. Biotinylated antibodies, horseradish peroxidase-conjugated streptavidin, and chromogenic substrates were used sequentially, and the microplates were then read on a microplate reader at 450 nm. The concentrations of the proinflammatory mediators were calculated using a standard curve plotted for each run.
Immunocytochemistry
Immunocytochemistry analysis was used to detect the expression of COX-2, iNOS, and the NF-κB p65 subunit in primary microglia according to the ABC kit procedure. The cells were cultured on coverslips in 24-well plates (1×105/well) and treated with LIG or LPS according to the experimental design. The immunoreactivity of the NF-κB p65 subunit and the inflammatory enzymes COX-2 and iNOS was examined 1 h or 24 h after LPS treatment, respectively. The cells were fixed with 4% paraformaldehyde for 30 min at 37 °C and washed with PBS for 5 min. The fixed cells were then permeabilized with 0.3% Triton X-100 in PBS for 30 min at 37 °C and washed with PBS for 5 min. The permeabilized cells were then rinsed with 3% H2O2 for 30 min to block endogenous peroxide activity and incubated with 5% bovine serum albumin from the ABC kit for 30 min to block nonspecific binding. The cells were then incubated with rabbit polyclonal antibody against NF-κB, COX-2, and iNOS (1:200), respectively, overnight at 4 °C. Primary antibodies were recognized by biotinylated anti-rabbit secondary antibody at 37 °C for 1 h and detected using the ABC kit at 37 °C for 1 h. Immunoreactions were visualized using 3,3'-diaminobenzidine tetrahydrochloride (DAB) as a chromagen. The cells detected for COX-2 and iNOS were then counterstained with hematoxylin to visualize nuclei. Negative controls were prepared identically, with the exception of the omission of primary antibodies. Photomicrographs were taken using an Olympus BX51 digital camera (Olympus Corporation, Shinjuku-ku, Tokyo, Japan). Three coverslips were used in each group, and the experiments were repeated twice. For quantitative analysis, the number of cells with p65 nuclear translocation or expression of COX-2 or iNOS in five random fields per coverslip were counted manually under a light microscope at 200×magnification. Immunoreactive microglia are expressed as a percentage of total cells counted in each coverslip.
Statistical analysis
Results are presented as mean±SD. The data were analyzed by one-way analysis of variance (ANOVA) using SPSS software (version 17.0). Values of P<0.05 were considered statistically significant.
Results
Effect of LIG on cellular viability
To avoid the potential cytotoxic effect of LIG, the MTT test was performed to determine the safe concentration range of LIG in primary rat microglia. The results showed that LIG at concentrations ranging from 2.5 to 20 μmol/L had no significant effect on microglial viability (P>0.05; Figure 2). These data showed that the results from the corresponding quantitative analysis were not influenced by the cell counts.

Effect of LIG on cellular viability of primary rat microglia treated with LPS. Primary rat microglia were pretreated for 1 h with LIG at the indicated concentrations (LIG 2.5, 5, 10, and 20 μmol/L) and then incubated with LPS (1 μg/mL) for an additional 24 h. Cell viability was examined by the MTT assay, and the results are expressed as a percentage of LPS treatment alone. The data from one representative experiment from three independent experiments are expressed as mean±SD (n=12).
LIG inhibits LPS-induced NO production
To study the anti-inflammatory effects of LIG in vitro, we investigated its effect on NO production in primary rat microglia. As shown in Figure 3, LPS stimulation for 24 h resulted in a marked induction of NO production compared with untreated control cells (P<0.01). However, LIG significantly and concentration-dependently reduced LPS-induced NO production. Pretreatment of microglia with LIG at 2.5, 5, 10, and 20 μmol/L for 1 h prior to LPS stimulation decreased NO production to 75.9%±6.6%, 54.4%±2.8%, 43.1%±1.3%, and 47.6%±8.7% (P<0.05 or P<0.01 vs LPS group), respectively, with maximal inhibitory efficacy at 10 μmol/L (P>0.05 vs 20 μmol/L group).

LIG decreased NO production in LPS-stimulated primary rat microglia. Primary rat microglia were pretreated for 1 h with LIG at the indicated concentrations (LIG 2.5, 5, 10, and 20 μmol/L) and then incubated with or without LPS (1 μg/mL) for an additional 24 h. The content of NO in the culture supernatant was measured using Griess reagent. NO production is expressed as a percentage of LPS treatment alone. The data are representative of results obtained from three independent experiments (mean±SD, n=3). bP<0.05, cP<0.01 vs LPS group.
LIG inhibits LPS-stimulated release of inflammatory mediators
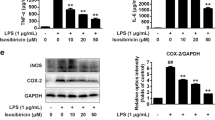
To study whether pro-inflammatory cytokines and chemokine factors are amenable to modulation by LIG in LPS-activated microglia, the levels of TNF-α, IL-1β, and MCP-1 in cell culture supernatants were measured with ELISA. LPS significantly elevated the production and release of TNF-α (167.4±13.1 pg/mL vs 2.1±1.8 pg/mL, P<0.01), IL-1β (33.0±7.5 pg/mL vs 0.0±0.0 pg/mL, P<0.01), and MCP-1 (2739±77 pg/mL vs 471±151 pg/mL, P<0.01) in primary rat microglia compared with control cultures. However, pretreatment with LIG at 2.5, 5, 10, and 20 μmol/L concentration-dependently reduced TNF-α content to 144.3±21.3 pg/mL, 114.4±18.5 pg/mL, 67.2±1.5 pg/mL, and 66.7±4.5 pg/mL, respectively (P<0.01, with the exception of 144.3±21.3 pg/mL at 2.5 μmol/L LIG; Figure 4A), IL-1β content to 10.4±3.1 pg/mL, 9.2±0.5 pg/mL, 0.2±0.4 pg/mL, and 0.0±0.0 pg/mL, respectively (P<0.01; Figure 4B), and MCP-1 content to 2290±263 pg/mL, 1358±295 pg/mL, 1243±213 pg/mL, and 1161±231 pg/mL, respectively (P<0.05 or P<0.01; Figure 4C). As shown in Figure 4D, LIG at 2.5, 5, 10, and 20 μmol/L concentration-dependently reduced the LPS-induced increase in the percentage of TNF-α to 86.2±12.7%, 68.3±11.0%, 40.1±0.9%, and 39.9±2.7% (P<0.01, with the exception of 86.2±12.7% at 2.5 μmol/L LIG), IL-1β to 31.5±9.3%, 27.7±1.6%, 0.6±1.3%, and 0.0±0.0% (P<0.01), and MCP-1 to 84.4±9.6%, 50.0±10.8%, 45.0±7.8%, and 42.0±8.4% P<0.05 or P<0.01), respectively. Consistent with the relationship of the concentration-response function of LIG on NO production in LPS-stimulated microglia, LIG tended to exhibit maximal inhibitory efficacy on the release of pro-inflammatory factors at 10 μmol/L (P>0.05 vs 20 μmol/L group).

LIG suppressed TNF-α, IL-1β, and MCP-1 production in LPS-stimulated primary rat microglia. Primary rat microglia were pretreated for 1 h with LIG at the indicated concentrations (LIG 2.5, 5, 10, and 20 μmol/L) and then incubated with LPS (1 μg/mL) for an additional 24 h. The levels of (A) TNF-α, (B) IL-1β, and (C) MCP-1 were measured in the culture media by specific ELISA. The data are representative of results obtained from three independent experiments (mean±SD, n=3). (D) The inhibitory effect of LIG on the production of inflammatory cytokines. Data are expressed as a percentage of LPS treatment alone. bP<0.05, cP<0.01 vs LPS group.
LIG suppresses LPS-induced activation of NF-κB and inflammatory enzyme expression
To further understand the effect of LIG on the inflammatory response in LPS-stimulated microglia, the immunoreactivity of the NF-κB p65 subunit and inflammatory enzymes COX-2 and iNOS were detected with immunocytochemistry 1 h or 24 h after LPS treatment, respectively. Figure 5A shows that NF-κB p65 subunit immunostaining was predominantly located in the cellular cytoplasm in the control group (a), which was mainly translocated into the nucleus following LPS stimulation (b). LIG (10 μmol/L) markedly prevented LPS-induced nuclear translocation of the NF-κB p65 subunit (c). Additionally, LPS induced most of the microglia exhibiting brown positive expression of COX-2 and iNOS (e and h), whereas few of the immunoreactive cells were detected in the control group (d and g). LIG (10 μmol/L) almost completely inhibited COX-2 and iNOS immunoreactivity in LPS-stimulated cells (f and i). Moreover, consistent with its inhibitory effects on the expression of these inflammatory enzymes, LIG treatment also effectively prevented LPS-induced morphological changes in microglia from the resting ramified form to the activated amoeboid form (Figure 5A). The quantitative analysis revealed that the percentage of microglia with activated NF-κB p65, COX-2, and iNOS was 0.5%±0.0%, 5.4%±0.5%, and 7.4%±0.3%, respectively, in the control cultures. LPS significantly increased the expression of activated NF-κB (94.5%±5.0%), COX-2 (97.3%±3.0%), and iNOS (95.0%±4.0%), which was significantly reduced to 16.2%±4.0%, 3.3%±2.0%, and 10.7%±2.0%, respectively, by pretreatment with 10 mol/L LIG (P<0.01 vs LPS group; Figure 5B).

LIG inhibited the activation of NF-κB and inhibited the expression of COX-2 and iNOS in LPS-stimulated primary rat microglia. Primary rat microglia were pretreated for 1 h with LIG at 10 μmol/L and then incubated with LPS (1 μg/mL) for the indicated times. The immunoreactivity of activated NF-κB and the inflammatory enzymes COX-2 and iNOS were detected with immunocytochemistry 1 h or 24 h following LPS treatment, respectively, among which the cells examined for inflammatory enzymes were further stained with hematoxylin. (A) Representative immunocytochemical photomicrographs of NF-κB (a–c), COX-2 (d–f), and iNOS (g–i) in primary rat microglia. In the control group (Con), the NF-κB p65 subunit was predominantly located in the cellular cytoplasm (a), which was mainly translocated into the nucleus by LPS within 1 h of stimulation (b). However, pretreatment for 1 h with LIG (10 μmol/L) markedly prevented LPS-induced nuclear translocation of the NF-κB p65 subunit (c). Additionally, few COX-2 and iNOS immunoreactive cells were detected in the resting ramified control cultures (d and g). LPS induced almost all microglia exhibiting brown positive expression of inflammatory enzymes accompanied by activated amoeboid morphology (e and h), which was significantly inhibited by 10 μmol/L LIG (f and i). (B) Quantification of immunoreactivity of activated NF-κB, COX-2, and iNOS in microglia by calculating the percentage of immunoreactive cells with regard to total cells counted in five representative areas on each coverslip. Data are expressed as mean±SD from two independent experiments (n=6). cP<0.01 vs LPS group.
Discussion
Our previous studies showed that LIG was capable of crossing the blood-brain barrier11 and significantly protected against cerebral ischemic damage and cognitive dysfunction, possibly via multiple mechanisms in rodent stroke and AD models4, 5, 6, 7, 8. Accumulating evidence suggests that the anti-inflammatory effects of LIG likely contribute to its neuroprotective action8, 12, 13. However, whether such anti-inflammatory effects of LIG result from the direct suppression of microglia, which are the only resident innate immune cells in the brain, has not been clear. Moreover, unknown are the other neuroinflammatory mediators that can be regulated by LIG during the inflammatory response. The present study, therefore, focused on the anti-inflammatory effects of LIG specifically on LPS-activated primary microglia in vitro. We found that LIG effectively prevented LPS-induced production of NO and proinflammatory cytokines (TNF-α, IL-1β, and MCP-1), expression of inflammatory enzymes (COX-2 and iNOS), and activation of NF-κB, as well as the activated morphological changes of primary microglia. Considering the anti-inflammatory effects of LIG in vivo8, the present study demonstrated that the neuroprotective effects of LIG were at least partially associated with significant inhibition of activated microglia-mediated neuroinflammation.
Microglia comprise about 10% of the total cell population of the brain but have multiple morphological and possible functional profiles that are influenced by their environment. The dual role of microglia has recently received much attention in NDD research14, 15. As the resting resident mononuclear phagocytes in the healthy brain, microglia are sensitive to environment changes and support endangered neurons or fend off threats to tissue integrity. For example, reactive microglia in the early stage of AD-like transgenic mouse models proved to be beneficial for the clearance of Aβ deposits and thus delayed AD progression. However, as the pathological process of AD developed, extended microglial activation exerted a significant reduction in the expression of their Aβ-binding receptors and Aβ-degrading enzymes, and thus led to phagocytotic activity dysfunction. However, the activated glia retained their ability to produce various pro-inflammatory mediators that induce the subsequent neuroinflammatory response16. Numerous studies have demonstrated that neuroinflammation may drive NDD pathogenesis. Nonsteroidal anti-inflammatory drugs effectively improved NDD outcome in preclinical and clinical trials17, 18. Therefore, blockade of the microglial activation-induced inflammatory response may have important therapeutic potential for the treatment of NDD and stroke, which are associated with excessive microglial activation19, 20.
LPS, a natural toll-like receptor 4 (TLR4) ligand, has been the most extensively utilized microglial activator for the induction of an inflammatory response. LPS binding protein works as a chaperon that enhances the binding of LPS to its intermediate receptor CD14. The association between the LPS-CD14 complex and TLR-4, together with the accessory adaptor protein MD2, initiates a plethora of downstream signaling events, including the transcription factor NF-κB pathway21. NF-κB is a heterodimeric transcription factor composed of p50 and p65 subunits from the Rel family of proteins. It is sequestered in the cytoplasm as an inactive complex when associated with its inhibitor, IκB. Upon stimulation with LPS, IκB is phosphorylated and degraded through proteosome-mediated pathways. The dissociated NF-κB p65 subunit then translocates into the nucleus and triggers the transcription of multiple genes, which leads to the production and release of proinflammatory cytokines (eg, TNF-α, IL-1β, and MCP-1), the induction of COX-2 and iNOS expression resulting in the biosynthesis and release of prostaglandins (PGs) and NO, and activation of NADPH oxidase-generating superoxide anions that combine with NO from iNOS to form the more damaging peroxynitrite (ONOO-) free radical10, 22, 23, 24. The collective insult by microglia-released cytokines, reactive oxygen species, and lipid metabolites eventually leads to neurotoxicity.
Consistent with numerous reports, our results showed that LPS mediated an intense proinflammatory response in primary rat microglia. The NF-κB p65 subunit translocated into the nucleus in >95% of microglia following 1 h stimulation with 1 μg/mL LPS. The production of NO free radicals and proinflammatory cytokines (eg, TNF-α, IL-1β, and MCP-1) and the expression of the inflammatory enzymes COX-2 and iNOS were subsequently increased in the microglia activated by LPS following 24 h treatment. LIG at concentrations ranging from 2.5 to 10 μmol/L, however, significantly and concentration-dependently suppressed the above microglia activation and proinflammatory response induced by LPS, without causing cytotoxicity. This is the first study to demonstrate the anti-inflammatory effects of LIG, which are likely attributable to inhibition of the TLR4/NF-κB-dependent pathway in microglia. The present results are consistent with a previous study showing the anti-inflammatory effects of LIG through inhibition of the Aβ5-35-induced NF-κB-dependent pathway in rat brains8. Altogether, our in vitro and in vivo studies indicate that the anti-neuroinflammatory effects of LIG at least partially involve inhibition of NF-κB signaling pathway activation. Furthermore, several cellular signal transduction pathways may be responsible for the inflammatory response in microglia, including the mitogen-activated protein kinase (MAPK) pathway21. Therefore, further studies are required to explore the overall molecular mechanisms of LIG against the neuroinflammatory response and subsequent neurotoxicity.
In summary, the present study demonstrated the potent inhibitory effects of LIG on the LPS-induced pro-inflammatory response in primary rat microglia. Our data suggest that the anti-inflammatory activity of LIG is mediated at least partially by its effects on the NF-κB signaling pathway. The present results provide valuable information about the mechanisms underlying the neuroprotective effects of LIG and the possible application of LIG for the treatment of neuroinflammatory diseases characterized by excessive microglial activation.
Author contribution
Jing WANG performed the research and analyzed the data. Jun-rong DU designed the project and wrote the paper. Xi KUANG contributed new analytical tools. Yu WANG and Cheng-yuan WANG helped for the experiment preparation.
References
Block ML, Hong JS . Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 2005; 76: 77–98.
Butovsky O, Talpalar E. A, Keren B. Y, Schwartz M. Activation of microglia by aggregated β-amyloid or lipopolysaccharide impairs MHC- II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Mol Cell Neurosci 2005; 29: 381–93.
Lin M, Zhu GD, Sun QM, Fang QC . Chemical studies of Angelica sinensis. Acta Pharm Sin 1979; 14: 529–34.
Yu Y, Du JR, Wang CY, Qian ZM . Protection against hydrogen peroxide-induced injury by Z-ligustilide in PC12 cells. Exp Brain Res 2008; 184: 307–12.
Kuang X, Yao Y, Du JR, Liu YX, Wang CY, Qian ZM . Neuroprotective role of Z-ligustilide against forebrain ischemic injury in ICR mice. Brain Res 2006; 1102: 145–53.
Kuang X, Du JR, Liu YX, Zhang GY, Peng HY . Postischemic administration of Z-ligustilide ameliorates cognitive dysfunction and brain damage induced by permanent forebrain ischemia in rats. Pharmacol Biochem Behav 2008; 88: 213–21.
Peng HY, Du JR, Zhang GY, Kuang X, Liu YX, Qian ZM, et al. Neuroprotective effect of Z-ligustilide against permanent focal ischemic damage in rats. Biol Pharm Bull 2007; 30: 309–12.
Kuang X, Du JR, Chen YS, Wang J, Wang YL . Protective effect of Z-ligustilide against amyloid β-induced neurotoxicity is associated with decreased pro-inflammatory markers in rat brains. Pharmacol Biochem Behav 2009; 92: 635–41.
Du J, Bai B, Kuang X, Yu Y, Wang C, Ke Y, et al. Ligustilide inhibits spontaneous and agonists- or K+ depolarization-induced contraction of rat uterus. J Ethnopharmacol 2006; 108: 54–8.
Akundi RS, Candelario-Jalil E, Hess S, Hüll M, Lieb K, Gebicke-Haerter PJ, et al. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia 2005; 51: 199–208.
Chen YY, Yu Y, Chen C, Yang W, Wang CY, Du JR . Pharmacokinetic profile of Z-ligustilide in rat plasma and brain following oral administration. Nat Prod Res Dev 2010; 22: 126–31.
Liu L, Ning ZQ, Shan S, Zhang K, Deng T, Lu XP, et al. Phthalide lactones from Ligusticum chuanxiong inhibit lipopolysaccharide-induced TNF-α production and TNF-α-mediated NF-κB activation. Planta Med 2005; 71: 808–13.
Du J, Yu Y, Ke Y, Wang C, Zhu L, Qian ZM . Ligustilide attenuates pain behavior induced by acetic acid or formalin. J Ethnopharmacol 2007; 112: 211–4.
Schwartz M, Butovsky O, Brück W, Hanisch UK . Microglial phenotype: is the commitment reversible? Trends Neurosci 2006; 29: 68–74.
Wyss-Coray T . Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 2006; 12: 1005–15.
Hickman ES, Allison EK, El Khoury J . Microglial dysfunction and detective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008; 28: 8354–60.
Jiang Q, Heneka M, Landreth EG . The role of peroxisome proliferators-activated receptor-γ (PPARγ) in Alzheimer's disease. CNS Drugs 2008; 22: 1–14.
Hirohata M, Ono K, Yamada M . Non-steroidal anti-inflammatory drugs as anti- amyloidogenic compounds. Curr Pharm Des 2008; 14: 3280–94.
Mosley RL, Gendelman HE . Control of neuroinflammation as a therapeutic strategy for amyotrophic lateral sclerosis and other neurodegenerative disorders. Exp Neurol 2010; 222: 1–5.
Lakhan SE, Kirchgessner A, Hofer M . Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 2009; 7: 97.
Medzhitov R, Preston–Hurlburt P, Janeway CA Jr . A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388: 394–7.
Thibeault I, Laflamme N, Rivest S . Regulation of the gene encoding the monocyte chemoattractant protein 1 (MCP-1) in the mouse and rat brain in response to circulating LPS and proinflammatory cytokines. J Comp Neurol 2001; 434: 461–77.
Yamada K, Komori Y, Tanaka T, Senzaki K, Nikai T, Sugihara H, et al. Brain dysfunction associated with an induction of nitric oxide synthase following an intracerebral injection of lipopolysaccharide in rats. Neurosci 1999; 88: 281–94.
Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, et al. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1–42- and lipopolysaccharide-activated microglia. J Neurosci 2002; 22: 3484–92.
Acknowledgements
The authors would like to thank Mr Yao YAO and Dr Eduardo CANDELARIO-JALIL for technical assistance with the primary microglial cultures.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, J., Du, Jr., Wang, Y. et al. Z-ligustilide attenuates lipopolysaccharide-induced proinflammatory response via inhibiting NF-κB pathway in primary rat microglia. Acta Pharmacol Sin 31, 791–797 (2010). https://doi.org/10.1038/aps.2010.71
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.71
Keywords
This article is cited by
-
A novel enzyme-assisted approach for efficient extraction of Z-ligustilide from Angelica sinensis plants
Scientific Reports (2017)
-
Combining Chemical Profiling and Network Analysis to Investigate the Pharmacology of Complex Prescriptions in Traditional Chinese Medicine
Scientific Reports (2017)
-
Study on effects of endophytes on growth and production of Z-Ligustilide and ferulic acid in Angelica sinensis
Brazilian Journal of Botany (2016)
-
Ligustilide Ameliorates Inflammatory Pain and Inhibits TLR4 Upregulation in Spinal Astrocytes Following Complete Freund’s Adjuvant Peripheral Injection
Cellular and Molecular Neurobiology (2016)
-
Neuroprotective effect of 2-(4-methoxyphenyl)ethyl-2-acetamido-2-deoxy-β-d-pyranoside against sodium nitroprusside-induced neurotoxicity in HT22 cells
Molecular and Cellular Biochemistry (2013)
