
Abstract
JFCR39 is an informatic anticancer drug discovery system that utilizes a panel of 39 human cancer cells coupled with a drug-activity database. This system not only provides disease-oriented information but can also predict the mechanism of action of a given antitumor agent. Development of a phosphatidylinositol 3-kinase (PI3K) inhibitor as an anticancer drug candidate has attracted a great deal of attention from both academia and industry because PI3K is known to be closely involved in carcinogenesis. ZSTK474 was identified as a PI3K inhibitor using JFCR39 system in combination with COMPARE analysis program. These findings were based on the similar fingerprint (growth inhibition profiles for JFCR39 human cancer cell line panel) with that of a classical PI3K inhibitor LY294002. Biochemical experiments confirmed ZSTK474 to be a potent pan-class I PI3K inhibitor, with high selectivity over other classes of PI3K and protein kinases. We previously reported the in vitro and in vivo antitumor efficacy of ZSTK474, together with the G0/G1 arrest and antiangiogenic activity. Here, we review the JFCR39 system and summarize recent studies on PI3K biology and the development of PI3K inhibitors before discussing ZSTK474 in some detail.
Similar content being viewed by others
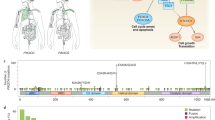


JFCR39 (Japanese Foundation for Cancer Research 39) system, an informatic anticancer drug discovery system for molecular target identification
JFCR39 is an informatic anticancer drug screening system we established in the early 1990s by utilizing a human cancer cell line panel JFCR39 coupled with a drug-activity database1, 2, 3, 4. This system was developed based on the NCI60 system5 with some modifications4. The JFCR39 system4 includes 30 cell lines in NCI60 together with 6 stomach cancer cell lines displaying high incidence in Japan, and 3 breast cancer cell lines (HBC-4, HBC-5, and BSY-1) established by the JFCR (Japanese Foundation for Cancer Research) (see Table 1).
One important function of JFCR39 is to provide disease-oriented information for personalized cancer chemotherapy, based on the differential growth inhibition activity of a certain antitumor agent against the 39 cancer cell lines. Inhibition of cell growth is assessed by the sulforhodamine B (SRB) assay, which determines the change in total cellular protein following 48 h of treatment with the antitumor agent1, 2, 6. The molar concentration of the agent required for 50% growth inhibition (GI50) of each cell in JFCR39 is then obtained1, 7, and the graphical representation (termed fingerprint) for the differential growth inhibition against the cells in the JFCR39 panel is finally plotted based on a calculation that uses a set of GI50 values8.
Another application of the JFCR39 system is to identify the mechanism of action or molecular target of an antitumor agent. The action mechanism of a drug candidate can be predicted by comparing its fingerprint with those of anticancer drugs or chemical tools with a known mechanism using the COMPARE algorithm, because the fingerprint represents the whole inhibition profiles of the related targets in the cells1, 4. The COMPARE analysis is performed by calculating the Pearson correlation coefficient (r) between the GI50 mean graphs of two compounds X and Y using the following formula: r=(∑(xi–xm) (yi–ym))/(∑(xi–xm)2 ∑(yi–ym)2)½, where xi and yi are Log GI50 of the two compounds, respectively, for each cell line, and xm and ym are the mean values of xi and yi, respectively (n=39)1, 8. The r value is then used to determine the degree of similarity, ie, the higher the r value is, the greater the similarity of X with Y. Generally, an r value of more than 0.5 between two agents suggests that two agents have a similar action mechanism, whereas a value of >0.8 suggests they have the same mechanism of action. Interestingly, we recently found that the difference of r value between 0.5 and 0.8 might reflect the different target specificity between two phosphatidylinositol 3-kinase (PI3K) inhibitors9.
So far, utilizing the JFCR39 system we have succeeded in predicting the action mechanisms of MS-247 (topoisomerase inhibitor), FJ5002 (telomerase inhibitor), ZSTK474 (PI3K inhibitor), and other antitumor compounds1, 10, 11. ZSTK474 is a very promising anticancer drug candidate that has been approved for clinical trials.
PI3K, a promising molecular target for cancer chemotherapy
PI3Ks are a family of lipid kinases that phosphorylate the 3′-OH of phosphoinositides (Figure 1)12, 13, 14. PI3Ks are divided into three classes based on their primary structure and in vitro substrate specificity15, 16. Class I PI3Ks are heterodimeric kinases as complexes of a catalytic subunit p110 with a regulatory subunit p85, p101, or p84. This class of PI3K preferentially phosphorylates PIP2 (phosphatidylinositol 4,5-bisphosphate) to generate PIP3 (phosphatidylinositol 3,4,5-trisphosphate) (Figure 1). Class II PI3Ks contain three members including PI3KC2α, PI3KC2β, and PI3KC2γ, which phosphorylate phosphatidylinositol (PI) and phosphatidylinositol 4-phosphate (PIP). This class of PI3K has no regulatory subunit and is known to be involved in membrane trafficking and receptor internalization17. Vacuolar protein sorting 34 (Vps34) is the sole Class III PI3K which phosphorylates PI to phosphatidylinositol 3-phosphate. Vps34 is known to play an important role in endocytosis and vesicular trafficking18, 19, 20, and has recently been reported to be essential for autophagy induction in response to nutrient availability20. Class I PI3Ks are often referred to simply as PI3Ks because they have been investigated far more than the other two classes. Phosphatidylinositol 4-kinases (PI4Ks) are a group of lipid kinases that phosphorylate PI to PIP at 4-OH. Mammalian PI4Ks were classified as types II and III based on their sensitivities to two specified inhibitors21. The so-called type I PI4Ks were later identified to be PI3K. By generating PIP, which is required for synthesis of PIP2, PI4Ks are also closely involved in cell signaling regulation, vesicular trafficking and endocytosis22. PI3K-related kinases (PIKKs), which are sometimes termed Class IV PI3Ks, are protein kinases with a similar structure to the catalytic subunits of PI3Ks. Examples of PIKKs include mTOR, DNA-dependent protein kinase (DNA-PK), and ataxia telangiectasia mutated gene product (ATM). These protein kinases are known to be involved in protein synthesis or DNA repair23.

Schematic structures of Class I, II, III PI3K, PI4K, and PTEN, and the related lipid reactions they catalyze. PI3K phosphorylates 3-OH of phosphoinositides. Class I PI3K preferentially phosphorylates PI(4,5)P2 to generate PI(3,4,5)P3. As a counterpart of class I PI3K, PTEN dephosphorylates PI(3,4,5)P3 to produce PI(4,5)P2. Class II PI3K phosphorylates PI(4)P and PI, to PI(3,4)P2 and PI(3)P, respectively. Class III PI3K phosphorylates PI to PI(3)P. In contrast to PI3K, PI4K phosphorylates PI at 4-OH to produce PI(4)P. PI: phosphatidylinositol; PI(4)P: phosphatidylinositol 4-phosphate; PI(3)P: phosphatidylinositol 3-phosphate; PI(4,5)P2: phosphatidylinositol 4,5-bisphosphate; PI(3,4,5)P3: phosphatidylinositol 3,4,5-trisphosphate.
Class I PI3Ks are further divided into subclasses IA and IB based on their regulatory subunit and upstream regulator14. Class IA PI3Ks are mainly activated by various receptor tyrosine kinases (RTKs) and Ras24. There are three isoforms in Class IA including PI3Kα, PI3Kβ, and PI3Kδ, with the respective p110 catalytic subunit bound to the p85 regulatory subunit. Class IB PI3Kγ, which consists of catalytic subunit p110γ and a regulatory subunit p101 or p84. PI3Kγ is mainly activated by G-protein-coupled receptors (GPCRs) such as chemokine receptors25, 26, 27. While the PI3Kα and PI3Kβ are expressed ubiquitously, PI3Kδ and PI3Kγ are mainly expressed in leukocytes28, 29. In particular, PI3Kα is known to play an important role in tumorigenesis because a high frequency of gain-of-function mutations and amplification of PIK3CA, which encodes p110α, has been found in human cancers30, 31, 32, 33, 34. Additionally, PI3Kα was found to be involved in insulin signaling and glucose metabolism35. PI3Kβ was reported to activate platelets, suggesting a role in the development of thrombotic diseases36. Recently, various reports showed that PI3Kβ predominantly contributed to PIP3 production in PTEN (phosphatase and tension homolog deleted on chromosome ten, the catalytic counterpart of PI3K) negative cancers, suggesting the key role of PI3Kβ in the tumorigenesis with PTEN inactivation37, 38. PI3Kδ and/or γ inactivation leads to a severely impaired immune system39, 40, and blocks the recruitment of neutrophils to the sites of inflammation41, 42, suggesting that these two isoforms are involved in the immune system and inflammation. As the counterpart of PI3K, PTEN dephosphorylates PIP3 to produce PIP2. Like PI3K, PTEN is also closely involved in cancer since frequent loss-of-function mutations were found in various human cancers43. In addition, PI3K mutation and PTEN inactivation were reported to cause resistance to cancer therapies targeting the RTKs44.
Thus, PI3K is thought to be an attractive target for cancer chemotherapy due to the high frequency in cancer with mutation and amplification of PI3Ks, inactivation of the counterpart PTEN, and mutation of RTKs25. As shown in Figure 2, after activation by RTKs or Ras, PI3K phosphorylates PIP2 to produce PIP3, which is reversed by PTEN. PIP3 binds the PH (pleckstrin homology)-domain-containing protein kinases such as Akt and PDK, to activate and recruit them to the plasma membrane. Besides the direct activation by PIP3, Akt can also be regulated by PDK and mTOR complex 2 (mTORC2). Activation of Akt promotes cell cycle progression by regulating GSK3 (glycogen synthesis kinase 3) and the downstream cyclin D1, and by blocking forkhead (FOXO)-mediated transcription of Cdk (cyclin dependent kinase) inhibitor p27. Akt also acts to maintain cell survival through inhibition of BAD (Bcl2-antagonist of cell death). Furthermore, Akt promotes cell growth by phosphorylation of the downstream mTOR complex 1 (mTORC1)45, which translates mRNAs to protein via the p70S6K-S6 and 4E-BP1-eIF4E pathways46. In addition, hypoxia-inducible factor 1α (HIF-1α) was reported to be up-regulated downstream of mTORC1, and therefore promotes angiogenesis by transcribing VEGF (vascular endothelial growth factor)47. However, phosphorylation of S6K negatively regulates insulin receptor substrate (IRS) and PI3K, leading to a feedback loop48, 49, 50. Thus, inhibition of mTORC1 activates upstream proteins such as PI3K and Akt51, and thereby attenuates the inhibition potency.

PI3K/Akt pathway involved in tumorigenesis. After activation by RTKs or Ras, PI3K catalyzes the phosphorylation of PIP2 to generate PIP3, which activates Akt and PDK. Akt can also be activated by PDK and mTOR complex 2 (mTORC2, rictor-mTOR), other than direct activation by PIP3. By increasing the level of cyclin D1, and reducing the level of Cdk (cyclin dependent kinase) inhibitor p27, Akt promotes the cell cycle progression. Akt also acts to maintain cell survival by phosphorylation of BAD and release of the anti-apoptotic protein Bcl-2. Furthermore, Akt controls cell growth by phosphorylation of the downstream mTOR complex 1 (mTORC1, raptor-mTOR), which promotes translation of mRNAs to synthesize protein via p70S6K-S6 and 4E-BP1-eIF4E pathways. In addition, HIF-1α is up-regulated downstream of mTORC1, and then promotes angiogenesis via enhancing transcription of VEGF. However, the mTORC1/S6K cascade negatively regulates IRS, which leads to a feedback loop. PDK: 3-Phosphoinositide-dependent protein kinase; GSK3: glycogen synthesis kinase 3; FOXO: forkhead; p70S6K: p70S6 kinase; 4E-BP1: 4E-binding protein 1; IRS: insulin receptor substrate; HIF-1α: hypoxia-inducible factor 1α; VEGF: vascular endothelial growth factor.
In recent years, development of PI3K inhibitors attracted a great deal of attention from both academia and industry. In particular, elucidation of the crystal structure of PI3Kγ52 and those of its complexes with LY294002 and wortmannin53 has facilitated new drug design and thus further accelerated the development of novel PI3K inhibitors. Presently, about a dozen novel PI3K inhibitors are being evaluated in clinical trials (Table 2). Most of these drug candidates are pan-PI3K isoform inhibitors, with the exception of CAL-101 (also named IC87114) as a PI3Kδ specific inhibitor. Among the pan-PI3K isoform inhibitors, some exhibit selectivity over mTOR and DNA-PK, such as GDC-0941, XL-147, and BKM-12014, 54, whereas others show no corresponding selectivity, such as GDC-0980, XL-765, BEZ235, and GSK-212645814, 55, 56 (Table 2). Among the novel PI3K inhibitors, GDC-0941 and BEZ235 are the most intensively studied. Both compounds showed favorable efficacy in vitro and in vivo on various cancers without any obvious toxicity23, 57, 58, 59, 60. In addition to the investigation of these compounds as single agents, combinations of PI3K inhibitors with other drug candidates that have different molecular targets were also reported. In deed, a combination of a PI3K inhibitor with a MEK inhibitor was found to give enhanced efficacy in various tumor types61, 62.
ZSTK474, a promising PI3K inhibitor identified using the JFCR39 system
ZSTK474 is an s-triazine derivative synthesized by Zenyaku Kogyo as an anticancer drug candidate together with more than 1500 other analogues63. Before its identification by the JFCR39 system, ZSTK474 showed promising antitumor efficacy, although the molecular target was unknown. In 2003, growth inhibitory activity of ZSTK474 was examined against the JFCR39 panel and its corresponding fingerprint was established. COMPARE analysis was then carried out by comparing the fingerprint of ZSTK474 with those of other antitumor drugs and chemical tools that have known molecular targets in the JFCR39 drug-activity database. Intriguingly, a high r value of 0.766 was found between the fingerprint of ZSTK474 and that of LY294002, a classical PI3K inhibitor11. Therefore, we predicted that ZSTK474 might also be a PI3K inhibitor.
Next, we investigated whether ZSTK474 directly inhibits the activity of PI3K. A novel non-radioactive assay method, known as homogenous time-resolved fluorescence (HTRF) assay, was utilized to measure the level of inhibition against human recombinant PI3Kα, β, δ, and γ. As a result, ZSTK474 inhibited all the 4 PI3K isoforms potently, with IC50 values of 16, 44, 5, 49 nmol/L for PI3Kα, β, δ, and γ, respectively, suggesting that it is a pan-PI3K inhibitor64 (Figure 3A). Docking analysis was performed by using the crystal structure of PI3Kγ-LY294002 as a model structure. As shown in Figure 3B, ZSTK474 binds with PI3K in the ATP-binding pocket. To demonstrate the mode of inhibition, the Lineweaver-Burk plots were developed based on a series of kinase reactions in the presence of various concentrations of ATP and ZSTK474. As shown in Figure 3C, the 1/v versus 1/[ATP] plots with different concentrations of ZSTK474 intersect on the 1/v axis, indicating that ZSTK474 competes with ATP in inhibiting PI3Kα. The same mode of inhibition is also exhibited for the other three PI3K isoforms64.

Mode of inhibition of ZSTK474 against class I PI3K. (A) IC50 of ZSTK474 against 4 isoforms of class I PI3K. (B) ATP binding pocket occupied by ZSTK474 for binding with PI3Kγ11. (C) Lineweaver-Burk plot analysis of inhibition of PI3Kα by ZSTK47464. The plots with various concentrations of ZSTK474 intersect on the 1/v axis. These results suggest that ZSTK474 competitively inhibits the binding of ATP to class I PI3K.
To investigate the specificity of ZSTK474 as a PI3K inhibitor, we examined its inhibition against 139 known protein kinases11. As a result, no potent inhibition was shown even at a high concentration of ZSTK474 (30 μmol/L). We also checked its activity against other members of the PI3K superfamily, including class II and III PI3K, PI4K, and PIKK such as mTOR and DNA-PK. ZSTK474 was found to be a class I-specific PI3K inhibitor, by displaying selectivity over mTOR, DNA-PK, and other PI3K superfamily members. This characteristic is similar to that of GDC-0941, but different from NVP-BEZ235, which showed more potent activity against mTOR and DNA-PK than against class I PI3K9, 65. We also examined the growth inhibition profiles of these PI3K inhibitors across the JFCR39 panel and compared their fingerprints. Interestingly, ZSTK474 showed a more similar fingerprint with GDC-0941 (r=0.863), compared to that with BEZ235 (r=0.67)9, consistent with our biochemical assay results regarding the PI3K superfamily inhibition. We also compared the fingerprint of ZSTK474 with that of other PI3K inhibitors such as PX866 (a pan-PI3K inhibitor), PI-103 (a pan-PI3K inhibitor) and TGX-221 (a specific PI3Kβ inhibitor). All the r values are more than 0.5. In the case of the fingerprint of PI3Kβ isoform specific PI3K inhibitor TGX221, no higher difference was shown between the fingerprints of TGX221 and ZSTK474, compared with those of PI-103 and ZSTK474, suggesting JFCR39 might not predict the isoform specificity of PI3K inhibitor.
In vitro, ZSTK474 inhibited the growth of 39 human cancer cell lines with a mean GI50 value of 0.32 μmol/L11, (Figure 4A) and blocked cell cycle progression at G0/G1 phase in various human cancer cells without obvious induction of apoptosis11, 66. The G0/G1 arrest effect might be attributed to inactivation of cyclin D1, enhanced expression of p27, and the following pRB dephosphorylation66. Moreover, ZSTK474 showed potent anti-angiogenic effect67. In vitro, ZSTK474 inhibited HIF-1α expression and VEGF production in RXF-631L cells, and blocked the proliferation, migration, and tube formation of HUVECs (human umbilical vein endothelial cells) (Figure 4B). In vivo, a significant reduction of microvessel number was observed in tumor tissues of ZSTK474-treated RXF-631L xenografts, compared with those of vehicle-treated controls. The in vivo anti-angiogenic effect is attributed to its dual inhibition mechanism: inhibition of VEGF secretion by cancer cells and direct inhibition of PI3K in endothelial cells67. Oral administration of ZSTK474 indicated favorable in vivo antitumor efficacy on various cancer xenografts at both early and advanced stages, without any obvious toxicity66, 67, 68 (Figure 4C). The expression of phospho-Akt (ser 473) correlates with antitumor efficacy, suggesting this could act as a predictive biomarker 68. Given the favorable preclinical antitumor effect and safety of ZSTK474, the FDA of USA has recently approved the evaluation of this drug candidate in phase I clinical trials.

In vitro and in vivo antitumor activities of ZSTK474. (A) Fingerprint of ZSTK474 for the JFCR39 panel9. Fingerprint indicates the differential growth inhibition pattern of ZSTK474 for the cell lines in JFCR39 panel. The X-axis shows difference in logarithmic scale between the mean of Log GI50 values for all 39 cell lines (MG-MID, expressed as 0 in the fingerprint) and the Log GI50 for each cell line in JFCR39 panel. Columns to the right of 0 indicate the sensitivity of the cell lines to a given compound and columns to the left indicate the resistance. MG-MID=mean of Log GI50 values for all 39 cell lines; Delta=difference between the MG-MID and the Log GI50 value for the most sensitive cell line; Range=difference between the Log GI50 values for the most resistant cell line and the most sensitive cell line. (B) In vitro antiangiogenic effect of ZSTK474. Upper panel: Wound healing assay shows ZSTK474 potently inhibits migration of HUVECs at 1 μmol/L; lower panel: ZSTK474 potently blocks the in vitro tube formation by HUVECs67. (C) In vivo antitumor efficacy of ZSTK474. Oral administration of ZSTK474 at 400 mg/kg to WiDr xenograft daily from day 0 to 26, except for days 6, 13, and 20, lead to obvious tumor growth inhibition. The pictures were taken on day 2811.
Discussion
As a bioinformatic drug discovery system, JFCR39 has made a significant contribution to drug development both in Japan and around the world69. Information from the JFCR39 system has facilitated the selection of many compounds for active development as anticancer drug candidates, some of which have entered clinical trials70. In particular, JFCR39 has become a well known platform for molecular-targeted anticancer drug discovery in Japan. We believe, with the support of JFCR39, the first molecular-targeted anticancer drug originally developed in Japan will be a reality.
An important issue in the development of PI3K inhibitors as antitumor drug candidates is whether dual inhibition of PI3K and mTOR is superior to specifically targeting PI3K. Dual inhibition is considered to enhance antitumor efficacy. However, dual inhibitors of PI3K and mTOR often further target DNA-PK, and are therefore thought to bring about unfavorable side-effect71. Thus far, the preclinical data do not indicate which type of inhibitor is advantageous overall, because both display favorable antitumor efficacy and safety. Indeed, the main companies involved in the development of PI3K inhibitors, including Novartis, Genentech, and Exelixis, are currently evaluating both types of PI3K inhibitors in clinical trials (Table 2). Hence, a definite answer regarding the most desirable type of PI3K inhibitor will have to await the result of clinical trials.
As a novel PI3K inhibitor, ZSTK474 has displayed favorable antitumor efficacy in preclinical experiments. Given the key role of PI3K in fundamental cellular functions, such as growth, one might predict that PI3K inhibition will inevitably lead to serious side effects. Nevertheless, PI3K inhibitors such as ZSTK474 have exhibited favorable results concerning the safety72, 73, 74, 75, 76. Notably, XL-147 has entered phase II clinical trials, suggesting its clinical safety. In conclusion, the ongoing clinical evaluation on these PI3K inhibitors is expected to furnish a new class of molecular targeted anticancer drugs.
References
Yamori T, Matsunaga A, Sato S, Yamazaki K, Komi A, Ishizu K, et al. Potent antitumor activity of MS-247, a novel DNA minor groove binder, evaluated by an in vitro and in vivo human cancer cell line panel. Cancer Res 1999; 59: 4042–9.
Nakatsu N, Nakamura T, Yamazaki K, Sadahiro S, Makuuchi H, Kanno J, et al. Evaluation of action mechanisms of toxic chemicals using JFCR39, a panel of human cancer cell lines. Mol Pharmacol 2007; 72: 1171–80.
Dan S, Tsunoda T, Kitahara O, Yanagawa R, Zembutsu H, Katagiri T, et al. An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res 2002; 62: 1139–47.
Yamori T . Panel of human cancer cell lines provides valuable database for drug discovery and bioinformatics. Cancer Chemother Pharmacol 2003; 52 Suppl 1: S74–9.
Shoemaker RH . The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006; 6: 813–23.
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82: 1107–12.
Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991; 83: 757–66.
Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst 1989; 81: 1088–92.
Kong D, Dan S, Yamazaki K, Yamori T . Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR39. Eur J Cancer 2010; 46: 1111–21.
Naasani I, Seimiya H, Yamori T, Tsuruo T . FJ5002: a potent telomerase inhibitor identified by exploiting the disease-oriented screening program with COMPARE analysis. Cancer Res 1999; 59: 4004–11.
Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, et al. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst 2006; 98: 545–56.
Kong D, Yamori T . Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci 2008; 99: 1734–40.
Toker A, Cantley LC . Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 1997; 387: 673–6.
Kong D, Yamori T . Advances in development of phosphatidylinositol 3-kinase inhibitors. Curr Med Chem 2009; 16: 2839–54.
Wymann MP, Pirola L . Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta 1998; 1436: 127–50.
Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 2001; 70: 535–602.
Carracedo A, Pandolfi PP . The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 2008; 27: 5527–41.
Murray JT, Panaretou C, Stenmark H, Miaczynska M, Backer JM . Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic 2002; 3: 416–27.
Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol 1999; 1: 249–52.
Backer JM . The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J 2008; 410: 1–17.
D'Angelo G, Vicinanza M, Di Campli A, De Matteis MA . The multiple roles of PtdIns(4)P -- not just the precursor of PtdIns(4,5)P2. J Cell Sci 2008; 121: 1955–63.
Balla A, Balla T . Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol 2006; 16: 351–61.
Marone R, Cmiljanovic V, Giese B, Wymann MP . Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 2008; 1784: 159–85.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994; 370: 527–32.
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB . Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4: 988–1004.
Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC . Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 2005; 30: 194–204.
Rommel C, Camps M, Ji H . PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 2007; 7: 191–201.
Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem 1997; 272: 19236–41.
Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA 1997; 94: 4330–5.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554.
Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 2005; 11: 2875–8.
Whyte DB, Holbeck SL . Correlation of PIK3Ca mutations with gene expression and drug sensitivity in NCI-60 cell lines. Biochem Biophys Res Commun 2006; 340: 469–75.
Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999; 21: 99–102.
Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64: 7678–81.
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006; 125: 733–47.
Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med 2005; 11: 507–14.
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008; 454: 776–9.
Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA 2008; 105: 13057–62.
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002; 297: 1031–4.
Del Prete A, Vermi W, Dander E, Otero K, Barberis L, Luini W, et al. Defective dendritic cell migration and activation of adaptive immunity in PI3Kgamma-deficient mice. EMBO J 2004; 23: 3505–15.
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000; 287: 1049–53.
Puri KD, Doggett TA, Douangpanya J, Hou Y, Tino WT, Wilson T, et al. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood 2004; 103: 3448–56.
Yuan TL, Cantley LC . PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27: 5497–510.
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA . Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat 2008; 11: 32–50.
Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428: 332–7.
Giles FJ, Albitar M . Mammalian target of rapamycin as a therapeutic target in leukemia. Curr Mol Med 2005; 5: 653–61.
Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH . Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem 2004; 279: 45643–51.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166: 213–23.
Hay N . The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005; 8: 179–83.
Vogt PK, Kang S . Kinase inhibitors: vice becomes virtue. Cancer Cell 2006; 9: 327–8.
Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65: 7052–8.
Walker EH, Perisic O, Ried C, Stephens L, Williams RL . Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 1999; 402: 313–20.
Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 2000; 6: 909–19.
Sellers WR . NVP-BKM120, a selective inhibitor of type I PI3 kinases. Proc AACR 2010; 101: DD01–4.
Friedman LS, Belvin M, Berry L, Berry M, Castanedo G, Edgar K, et al. A novel potent and selective inhibitor of PI3K/mTOR, GDC-0980, currently in phase I clinical trials. Mol Cancer Ther 2009; 8: C201.
Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Identification of GSK2126458, a highly potent inhibitor of phosphoinositide 3-kinase (PI3K) and the mammalian target of repamycin (mTOR). Mol Cancer Ther 2009; 8: C62.
Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther 2009; 8: 1725–38.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin -4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 2008; 51: 5522–32.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008; 7: 1851–63.
McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res 2009; 69: 5835–42.
Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 2009; 15: 4649–64.
Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA 2009; 106: 19503–8.
Yaguchi S, Izumisawa Y, Sato M, Nakagane T, Koshimizu I, Sakita K, et al. In vitro cytotoxicity of imidazolyl-1,3,5-triazine derivatives. Biol Pharm Bull 1997; 20: 698–700.
Kong D, Yamori T . ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci 2007; 98: 1638–42.
Kong D, Yaguchi S, Yamori T . Effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor, on DNA-dependent protein kinase. Biol Pharm Bull 2009; 32: 297–300.
Dan S, Yoshimi H, Okamura M, Mukai Y, Yamori T . Inhibition of PI3K by ZSTK474 suppressed tumor growth not via apoptosis but G0/G1 arrest. Biochem Biophys Res Commun 2009; 379: 104–9.
Kong D, Okamura M, Yoshimi H, Yamori T . Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur J Cancer 2009; 45: 857–65.
Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, et al. Correlating Phosphatidylinositol 3-Kinase Inhibitor Efficacy with Signaling Pathway Status: In silico and Biological Evaluations. Cancer Res 2010; 70: 4982–94.
Kong D, Yamazaki K, Yamori T . Discovery of phosphatidylinositol 3-kinase inhibitory compounds from the SCADS (screening committee of anticancer drugs) library. Biol Pharm Bull 2009; 32: 297–300.
Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 2004; 57: 188–96.
Carnero A . Novel inhibitors of the PI3K family. Expert Opin Investig Drugs 2009; 18: 1265–77.
LoRusso P, Sarker D, Hoff DV, Tibes R, Derynck MK, Ware JA, et al. Pharmacokinetics and pharmacodynamic biomarkers for the pan-PI3K inhibitor GDC-0941: Initial Phase I evaluation. Eur J Cancer 2008; 6: 70s.
Markman B, LoRusso PM, Patnaik A, Heath E, Laird AD, van Leeuwen B, et al. A phase I dose-escalation study of the safety, pharmacokinetics and pharmacodynamics of XL765, a novel inhibitor of PI3K and mTOR, administered orally to patients with solid tumors. Eur J Cancer 2008; 6: 68s.
Calvo E, Edelman G, Baselga J, Kwak E, Scheffold C, Nguyen L, et al. A phase I dose-escalation study of the safety, pharmacokinetics and pharmacodynamics of XL147, a novel PI3K inhibitor administered orally to patients with advanced solid tumors. Eur J Cancer 2008; 6: 69s.
Wagner AJ, Burris HA, de Bono JS, Jayson GC, Bendell JC, Gomez-Roca C, et al. Pharmacokinetics and pharmacodynamic biomarkers for the dual PI3K/mTOR inhibitor GDC-0980: Initial phase I. Mol Cancer Ther 2009; 8: B137.
Faulkner N, LoRusso PM, Guthrie T, Soria, JC, Bahleda R, Bendell JC, et al. A phase 1 safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination wiht erlotinib in patients with advanced solid tumors. Mol Cancer Ther 2009; 8: C197.
Acknowledgements
We thank Dr RH SHOEMAKER and Dr KD PAULL for discussion on the establishment of JFCR39 anticancer drug screening system. This work was supported by a grant from the National Institute of Biomedical Innovation, Japan to Dr T YAMORI (05–13); a grants-in-aid of the Priority Area “Cancer” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to Dr T YAMORI (20015048); a grants-in-aid for Scientific Research (A) from Japan Society for the Promotion of Science to Dr T YAMORI (22240092); and a grant from Kobayashi institute for innovative cancer chemotherapy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kong, Dx., Yamori, T. ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor identified using the JFCR39 drug discovery system. Acta Pharmacol Sin 31, 1189–1197 (2010). https://doi.org/10.1038/aps.2010.150
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.150
Keywords
This article is cited by
-
Class I PI3K inhibitor ZSTK474 mediates a shift in microglial/macrophage phenotype and inhibits inflammatory response in mice with cerebral ischemia/reperfusion injury
Journal of Neuroinflammation (2016)
-
Neurotrophins regulate ApoER2 proteolysis through activation of the Trk signaling pathway
BMC Neuroscience (2014)
-
Synthesis and Antimetastatic Effect of E-salignone
Chemistry of Natural Compounds (2014)
-
In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA
Investigational New Drugs (2014)
-
A pharmacological model reveals biased dependency on PI3K isoforms for tumor cell growth
Acta Pharmacologica Sinica (2013)
