
Abstract
Nicotinic acetylcholine receptors (nAChRs) are expressed in brainstem and spinal cord regions involved in the control of breathing. These receptors mediate central cholinergic regulation of respiration and effects of the exogenous ligand nicotine on respiratory pattern. Activation of α4* nAChRs in the preBötzinger Complex (preBötC), an essential site for normal respiratory rhythm generation in mammals, modulates excitatory glutamatergic neurotransmission and depolarizes preBötC inspiratory neurons, leading to increases in respiratory frequency. nAChRs are also present in motor nuclei innervating respiratory muscles. Activation of post- and/or extra-synaptic α4* nAChRs on hypoglossal (XII) motoneurons depolarizes these neurons, potentiating tonic and respiratory-related rhythmic activity. As perinatal nicotine exposure may contribute to the pathogenesis of sudden infant death syndrome (SIDS), we discuss the effects of perinatal nicotine exposure on development of the cholinergic and other neurotransmitter systems involved in control of breathing. Advances in understanding of the mechanisms underlying central cholinergic/nicotinic modulation of respiration provide a pharmacological basis for exploiting nAChRs as therapeutic targets for neurological disorders related to neural control of breathing such as sleep apnea and SIDS.
Similar content being viewed by others

Introduction
Respiration is a vital behavior, essentially continuous from birth to death. Respiratory rhythm is generated within the central nervous systems (CNS). The coordinated motor patterns of the respiratory muscles including the pump, eg, diaphragm, intercostal and abdominal, muscles as well as the upper airway, eg, genioglossal, muscles involve multilevel and diverse neurotransmitter regulation.
ACh plays an important role in the neural control of respiration1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, including in central chemosensitivity, ie, the ability of the brain to sense CO2 (and/or pH) to regulate ventilation15, 16, 17, 18, 19, 20, 21. Impairments in central cholinergic system are implicated in pathophysiology of some prevalent neurological disorders involving respiratory control such as SIDS and sleep apnea13, 22, 23, 24, 25, 26. Studies of central cholinergic regulation of respiration date to the early 1930s. Intraventricular injection of ACh into the lateral ventricle or the third ventricle of the brain in anesthetized, vagotomized cats, usually induces temporary depression of breathing, but in some cases, produces acceleration1. Cerebral arterial injection or intraventricular application to the fourth ventricle of ACh increases the frequency and amplitude of respiration in dogs. These effects are potentiated by the acetylcholinesterase inhibitor physostigmine and persist after denervation of the carotid and aortic chemoceptors, confirming central components of these ACh effects2. Perturbations of ACh synthesis, release, degradation, or activation of ACh receptors, in the medulla result in perturbations of respiratory pattern both in vivo7, 8, 27 and in vitro6, 9, 10, 12, 19, 28, 29.
Pontine cholinergic mechanisms associated with state-dependent modulation of respiratory control have been recently reviewed30, 31. In this review, we focus on the role of nAChRs in the preBötC, an essential site for respiratory rhythm generation32, 33, 34, and in brainstem and spinal cord respiratory motor nuclei in regulation of respiration.
nAChRs have attracted wide research interests since, in addition to mediating endogenous cholinergic regulation of respiratory pattern, nAChRs mediate the effects of nicotine from tobacco smoke, a matter of considerable public health significance. We also outline recent research on the effects of perinatal exposure to nicotine on neonatal control of breathing. In the interest of focus, we do not discuss the role of nAChRs in central chemoreception.
Cholinergic innervation and nAChRs in brainstem and spinal cord regions involved in respiratory control
Respiratory rhythm is generated in the brainstem. Our current view of respiratory rhythm generation in mammals is that there are two distinct, normally coupled, rhythm generators. The primary site, the preBötC, is ventral to the nucleus ambiguus, midway between the facial nucleus and the obex, caudal to the Bötzinger Complex and rostral to the ventral respiratory group (VRG) in the rostroventrolateral medulla32, 33. The preBötC is postulated to generate inspiratory rhythmic activity that dominates breathing in mammals at rest34, 35. A second site that is postulated to drive active expiratory activity is close to the facial nucleus in the region of the retrotrapezoid nucleus/parafacial respiratory group (RTN/pFRG)34, 36. Respiratory rhythm is transmitted via medullary premotoneurons37, 38, 39 to respiratory motoneurons in the ventral horn of the spinal cord, including the phrenic nucleus, and medullary cranial motonuclei such as the XII nucleus; these motoneurons drive the muscles of the respiratory pump and those regulating airway resistance.
In the brainstem, the principal cholinergic projection system originates in the pontomesencephalic tegmental cholinergic complex that includes the pedunculopontine tegmental nucleus and the laterodorsal tegmental nucleus (PPT/LDT)40, 41. Cholinergic neurons in the PPT/LDT have descending projections to the medullary reticular nuclei and the lateral reticular nucleus40, 41, probably including the preBötC. Cholinergic neurons in the PPT/LDT also project to motor nuclei of cranial nerves including XII nucleus40, 41, 42. Numerous cholinergic neurons are found in the medullary reticular formation and near the ventral medullary surface43, 44, 45. The preBötC probably also receives cholinergic input from these local sources44.
XII motoneurons have cholinergic innervation. The terminals that make synaptic contact with XII motoneurons contain both choline acetyltransferase(ChAT) and acetylcholinesterases (AChEs)46, 47. The soma and proximal dendrites of XII motoneurons are covered by a plexus of large puncta expressing a vesicular acetylcholine transporter (VAChT)45, 48, 49, 50 and a high-affinity choline transporter51. Premotor neurons in the caudal medullary intermediate reticular (IRt) region provide inspiratory drive to XII motoneurons37, 38, 39, 52. Although inspiratory drive is mainly glutamatergic, over half of these premotor neurons retrogradely labeled from the XII nucleus express mRNA for ChAT and are ChAT immunoreative suggesting that medullary IRt neurons contribute to cholinergic inputs to XII motoneurons39.
Cholinergic synapses are found on the cell bodies and dendrites of spinal ventral horn motoneurons probably including phrenic motoneurons41, 53, and these synaptic terminals contain VAChT45, 48, 49, 50 and high-affinity choline transporter51. These synaptic terminals most likely derive from cholinergic neurons in the spinal cord with some from axon collaterals of motoneurons within the motor nuclei41, 53.
α4, α5, and β2 nAChR subunit mRNAs are found in the ventrolateral medulla and the spinal cord ventral horn in adult rats. In the spinal cord ventral horn, α2, α3, and α7 subunit mRNAs are also detected54, 55, 56. In the XII motor nucleus, α4 and β2 subunit mRNAs are found55.
Immunohistochemical studies using antibodies against nAChR subunits suggest that α7 subunits are expressed in the ventrolateral medulla, the XII nucleus and the ventral horn of the spinal cord57. α7 immunoreactivity is found in phrenic motoneurons and in preBötC neurokinin-1 receptor containing neurons58. α7, α4, and β2 subunit immunoreactivity is present on XII motoneurons59, 60.
Many of the above studies used nAChR subunit antibodies. These antibodies may lack specificity, as the antibodies against the α3-, α4-, α7-, β2-, and β4-nAChR subunits are immunoreactive on brain tissue of respective knock-out mice, lacking α3-, α4-, α7-, β2-, or β4-nAChR subunits. Thus, particular caution should be exerted in interpretation of data obtained with the standard protocols for immunohistochemistry and Western blotting in the brain using these antibodies61, 62.
Activation of nAChRs in the preBötC modulates respiratory rhythm
In early studies, ionophoretic administration of cholinergic agents revealed the effects of ACh on respiratory neurons recorded extracellularly in ventral medulla in adult mammals in vivo. However, it was controversial whether nAChRs contribute to regulation of respiratory circuits and whether activation of nAChRs is excitatory or inhibitory63, 64, 65, 66, 67, probably due to the variability (or lack of precise information) concerning recording locations and the states of anesthesia68. Bilateral injection of nerve agent sarin, a potent acetylcholinesterase inhibitor, into a compact area near the lateral reticular nucleus in the medulla of rabbit causes respiratory arrest, which can be reversed by atropine69. This area of the rabbit is equivalent to the region we now define as the preBötC in the rat32, 33, the cat70, 71 and the rabbit72, 73, suggesting that the preBötC is a critical area where ACh acts to affect respiratory rhythm.
Bath application of ACh in the en bloc brainstem-spinal cord preparation from neonatal rat in vitro increases the frequency of respiratory rhythmic activity recorded from the 4th or 5th cervical spinal nerve (C4 or C5) ventral roots; this effect is diminished, but not completely abolished, by atropine. The effect of ACh can be completely abolished by further addition of the nicotinic antagonist dihydro-β-erythroidine (DHβE)6.
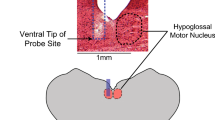
Unilateral microinjection of nicotine into the preBötC in the medullary slice preparation from neonatal rats that generates respiratory rhythm in vitro32 increases respiratory frequency and decreases the amplitude of rhythmic motor activity recorded from hypoglossal nerve root (XIIn). In contrast, nicotine injected into the XII nucleus induces tonic activity and an increase in amplitude but not in frequency of inspiratory bursts from XIIn. These effects can be blocked by the nicotinic antagonist mecamylamine (Figure 1). The results suggest that nicotine acts on the preBötC modulating respiratory frequency and on the XII nucleus to modulate the amplitude of inspiratory bursts28.

Unilateral microinjection of nicotine into the preBötC increases frequency and decreases amplitude of respiratory-related rhythmic activity. Rhythmic activity was recorded from hypoglossal nerve roots (XIIn) in the medullary slice preparation in vitro and the signal was integrated. Left panel: Microinjection (↑) of 10 nL 20 μmol/L nicotine into: 1, ipsilateral preBötC; 2, contralateral preBötC; 3, ipsilateral hypoglossal (XII) nucleus and 4, contralateral XII nucleus respectively. Injection pipettes were inserted into the loci 100−200 μm below the surface of the slice. Right panel: bath applied 1 μmol/L mecamylamine (Meca) blocked nicotine-induced responses. Reproduced from reference 28.
Bath application of low concentrations of nicotine (0.2−0.5 μmol/L, as low as the arterial blood nicotine concentration after smoking a cigarette74) to rhythmic slices increases respiratory frequency in a concentration-dependent manner. In voltage-clamped preBötC inspiratory neurons, nicotine induces a tonic inward current associated with an increase in membrane noise, increases the frequency and amplitude of spontaneous EPSCs during the expiratory period as well as decreases the amplitude of phasic inspiratory drive currents. These effects can be blocked by mecamylamine. These results indicate that nicotine differentially modulates excitatory neurotransmission in the preBötC: it enhances tonic excitatory input to, and inhibits excitatory coupling between, preBötC inspiratory neurons28. Based on a computational model of rhythmogenesis75, these cellular mechanisms can account for the nicotinic effects on respiratory frequency and pattern28. Since miniature EPSC analyses have not been performed, pre- and/or post synaptic mechanisms of nicotinic modulation of glutamatergic transmission and modulation of the excitability of preBötC inspiratory neurons remain to be determined. In addition to the classical synaptic transmission, cholinergic modulation of the excitability of preBötC inspiratory neurons and respiratory pattern may act via volume transmission mechanisms76.
The α7 nAChR antagonists α-bungarotoxin or methyllycaconitine (MLA) have little effect on the actions of low concentrations of nicotine on preBötC inspiratory neurons and respiratory pattern in the medullary slice preparation. In contrast, DHβE or hexamethonium completely reverse these nicotinic actions, which are also reduced by d-tubocurarine. Comparable concentrations of RJR-2403, an agonist selective for α4β2 nAChRs77, have effects similar to those of nicotine. MLA has little effect on RJR-2403 actions, while DHβE completely reverses them29. These pharmacological characteristics suggest that the predominant subtype of preBötC nAChRs that mediates the modulation of respiratory pattern by low concentrations of nicotine is an α4β2* combination and not an α7 subunit homomer. Results of pharmacological studies78 using the en bloc brainstem-spinal cord preparation from neonatal rats where respiratory motor activity can be recorded from C4 nerve roots are basically consistent with those of slice preparations. DHβE hyperpolarizes and decreases intraburst firing frequency in both inspiratory and preinspiratory (pre-I) neurons in the rostral ventrolateral medulla. MLA has no effects on the membrane potential of inspiratory neurons but hyperpolarizes and decreases intraburst firing frequency in pre-inspiratory neurons78. These authors conclude that α4β2 nAChRs mediate cholinergic modulation of both inspiratory and pre-inspiratory neurons whereas α7 nAChRs are only involved in cholinergic modulation of pre-inspiratory neurons. However, the concentrations of DHβE and MLA (20 μmol/L) used in this study were too high to be pharmacologically specific for nAChR subtypes. Such high concentrations may produce non-specific effects, calling into question their conclusions regarding the role of specific nAChR subtypes.
Transgenic nAChR subunit knock-out and knock-in mice provide a powerful approach to determine the molecular composition of nAChRs and their role in CNS functions. A knock-in mouse strain with a leucine to alanine mutation in the M2 pore-lining region (L9'A) of the nAChR α4 subunit renders α4-containing receptors hypersensitive to agonists79. In homozygous L9'A knock-in mice compared with wild-type mice, nicotine affects respiratory rhythm at ∼100-fold lower concentrations. Figure 2 shows that nicotine at low nanomolar concentrations (5 nmol/L, bath applied) depolarizes preBötC inspiratory neurons and increases respiratory frequency in L9'A mouse slices; these effects are blocked by DHβE (0.2 μmol/L). Responses of preBötC inspiratory neurons of L9'A mice to nicotine at low nanomolar concentrations are similar to those in wild-type mice at micromolar concentrations. These responses include: i) tonic inward current associated with an increase in membrane noise; ii) decrease in phasic inspiratory drive current; iii) increase in frequency and amplitude of spontaneous EPSCs, and; iv) increase in respiratory frequency and tonic/seizure-like activity in XIIn rhythmic motor output. These effects can be blocked by the α4* nAChRs selective antagonist DHβE. These data showing nicotine hypersensitivity of nAChRs in preBötC inspiratory neuron in L9'A mice indicate that these nAChRs contain α4 subunits. Nicotine facilitates glutamatergic neurotransmission and increases the excitability of preBötC inspiratory neurons via α4* nAChRs80. In resting conditions (without nicotine), the respiratory frequency of L9'A mouse slices is higher than that of wild-type slices suggesting that the preBötC nAChRs are hypersensitive to endogenously released ACh81. Unilateral microinjection of 50 nmol/L nicotine into the preBötC increases respiratory frequency. Microinjection of nicotine into the XII nucleus induces tonic/seizure-like activity in the ipsilateral XIIn. Bath application of DHβE blocks these effects. These results suggest that functional α4* nAChRs are present in both the preBötC and the XII nucleus. However, the effects of activating these receptors differ: α4* nAChRs in the preBötC mediate ACh/nicotinic modulation of respiratory rhythm, and α4* nAChRs in the XII nucleus mediate ACh/nicotine modulation of tonic activity in XIIn80.

Nicotine (5 nmol/L, bath applied) depolarizes preBötC inspiratory neurons and increases respiratory frequency in L9'A mouse slices; the effects are blocked by DHβE (0.2 μmol/L). Simultaneous whole-cell current-clamp recording from an inspiratory neuron (upper trace) in the preBötC and respiratory-related rhythmic activity from the XIIn (∫XII: integrated nerve activity) in medullary slice from L9'A mice. The neuron fired bursts of action potentials on top of rhythmic inspiratory depolarization synchronized with the rhythmic motor activity of XIIn. Insets: activities of both channels on an expanded time scale at time sections indicated. Time scales for all insets are equivalent. Y-scales for the continuous recordings and all the insets are equivalent for corresponding channels. Reproduced, with permission of J Neurosci, from reference 80.
Inhibition of AChEs by physostigmine produces both muscarinic and nicotinic effects in the medullary slice preparation. Physostigmine enhances the excitability of preBötC inspiratory neurons, induces tonic activity and an increase in frequency, amplitude and duration of inspiratory bursts of XIIn motor output. This suggests that endogenous ACh modulates excitatory neurotransmission and the excitability of preBötC respiratory neurons that, in turn, regulates respiratory frequency and pattern82.
Activation of nAChRs in respiratory motor nuclei modulates the pattern of rhythmic and tonic activity of motor output
Respiratory motoneurons are cholinergic. They release ACh at neuromuscular junctions. Respiratory drive transmitted to these motoneurons is glutamatergic, mediated primarily by non-NMDA receptors83, 84. Respiratory motoneurons also receive cholinergic inputs40, 41, 42, 45, 48, 49, 50, 51 that play modulatory role.
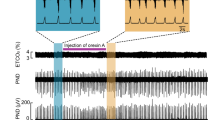
Motoneurons in the XII nucleus innervate the tongue and upper airway muscles85, 86. They have respiratory-related rhythmic activity and play an important role in regulating upper airway resistance and patency87. Activation of nAChRs in XII motoneurons excites these neurons28, 88, 89, 90. In the medullary slice preparation from neonatal rats in vitro, the nicotinic agonist, 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP) induces an inward current associated with an increase in membrane conductance in voltage-clamped XII motoneurons in the presence of TTX and atropine. This current is concentration-dependent (Figure 3) and blocked by DHβE. These results suggest that cholinergic/nicotinic modulation of the excitability of XII motoneurons is via post- and/or extra-synaptic nAChRs that do not contain the α7 subunit89. Microinjection of low nanomolar concentrations of nicotine into the XII nucleus in medullary slices from the mutant nAChR α4 subunit L9'A mice induces tonic/seizure-like activity in the XIIn. These effects are blocked by DHβE80. These results are consistent with Zaninetti et al(1999)88 and Chamberlin et al (2002)89 and suggest that the nAChRs in the XII nucleus contain α4 subunits.

1,1-dimethyl-4-phenylpiperazinium iodide (DMPP, a nicotinic agonist) produces inward currents that are dose-dependent and associated with an increase in membrane conductance in XII motoneurons. (A) An inward current in a voltage-clamped (-60 mV) XII motoneuron induced by bath-applied 25 μmol/L DMPP (bar). *I/V curve was measured during the break in record. (B) Peak amplitudes of DMPP-induced current (IDMPP) as a function of concentration. Atropine (1 μmol/L) was presence in the bath solution for (A) and (B). Reproduced from reference 89.
In a medullary slice preparation, bath application of 0.5 μmol/L nicotine decreases the frequency, but not the amplitude, of glutamatergic miniature EPSCs in XII motoneurons. Further time course analysis suggests that activation of presynaptic nAChRs usually facilitates glutamatergic synaptic transmission to XII motoneurons and then depresses it probably via receptor desensitization60.
In anesthetized, vagotomized adult rats in vivo, microdialysis of the nicotinic receptor agonist DMPP into the XII nucleus increases tonic and respiratory-related activity of the genioglossus muscle91. These studies suggest that nAChRs can be a potential therapeutic target for treatment of obstructive sleep apnea92, 93 that involves sleep-related loss of tone in the genioglossus muscle.
Studies on cholinergic regulation of phrenic motoneurons are scarce. Intrathecal injection of ACh at the C4 spinal segment increases the inspiratory activity of the phrenic nerve in anesthetized, immobilized and vagotomized rabbits94. However, microinjection of carbachol into the phrenic nucleus decreases the inspiratory activity of the phrenic nerve in anesthetized, immobilized rats95. These apparently contradictory observations may be due to species differences. Both the excitatory effects94 and inhibitory effects95 can be blocked by muscarinic ACh receptor antagonists. The functional significance of the nAChRs in the phrenic nucleus revealed by anatomical approaches58 awaits investigation.
Effects of perinatal nicotine exposure on neonatal respiratory control
SIDS is a leading cause of infant death between one month and one year of age, resulting in 0.55 deaths per 1000 live births in the United States96. The cause of SIDS is unknown. Impaired cadiorespiratory control and arousal responsiveness are hypothesized to be important mechanisms97, 98. Prone sleeping position and perinatal exposure to cigarette smoke are the leading risk factors for SIDS. Since the successful “back to sleep” campaign, cigarette smoke exposure during pre- and post-natal life has become the principal risk factor for SIDS99, 100.
Maternal smoking is associated with a trend of increased nicotine receptor binding in the brain in control groups of infants. However in SIDS infants (infants that died of SIDS) who were exposed to maternal smoking in utero, upregulation of nAChRs is absent in three brainstem nuclei related to arousal and cardiorespiratory control: the Nucleus Parabrachialis Lateralis, Locus Coeruleus and Nucleus Pontis Oralis101. These results suggest that altered development of nAChRs in brainstem cardiorespiratory and/or arousal circuits put some infants, i.e. those exposed to cigarette smoke in utero, at risk for SIDS.
In animal studies, prenatal exposure to nicotine affects neuronal development and upregulates nAChRs in the brain102, 103, 104, 105. Prenatal exposure to nicotine impairs protective responses to hypoxia in an age-dependent manner in rat pups106. Prenatal nicotine exposure alters the postnatal development of the ventilatory pattern and increases frequency of apneas107. Newborn mice prenatally exposed to nicotine exhibit unstable breathing and impaired arousal. Remarkably similar deficits are detected in pups lacking β2-containing nAChRs. Loss-of-function of these nAChRs reproduces many of the abnormalities caused by perinatal nicotine exposure suggesting that nicotine's detrimental side effects on a range of crucial defensive reflexes involve loss of function of nAChR subtypes, possibly via activity-dependent desensitization108.
Prenatal nicotine exposure delays early postnatal changes in breathing pattern in the neonatal mice. During normoxia, neonatal mice exhibit a high frequency of apnea [f(A)] that declines by postnatal day 3 (P3) in control animals, but persists in P3 nicotine-exposed mice. Hypoxia induces a rapid and sustained reduction in f(A). During recovery, f(A) increases above control levels. By P3 this increase is reduced in control but persists in nicotine-exposed mice. Nicotine locally applied over the XII motor nucleus in medullary slices from control neonatal mice in vitro increases tonic discharge and potentiates inspiratory burst amplitude in XIIn. In slices from prenatally nicotine-exposed mice, these effects are significantly reduced (Figure 4) suggesting that prenatal exposure to nicotine reduces cholinergic/nicotinic modulation of XII motoneurons90.

Prenatal nicotine exposure reduces response to nicotine receptor activation in hypoglossal (XII) nucleus. (A) Respiratory-related rhythmic activity from the XIIn (∫XII, integrated nerve activity) in control and prenatally nicotine-exposed medullary slices from P3 mice showing activity before, during and after 30 s local application of 10 μmol/L nicotine onto ipsilateral XII nucleus. Nicotine-mediated potentiation of XIIn inspiratory burst amplitude and tonic activity was diminished in slices from prenatally nicotine-exposed mice. (B) Respiratory-related rhythmic activity of XIIn from a control slice illustrates response to a 30 s application of 100 μmol/L nicotine before, during and after addition of 500 μmol/L nAChR antagonist hexamethonium bromide to the bathing solution. Reproduced, with permission of Blackwell Publishing Ltd, from reference 90.
Bath application of the GABAA receptor agonists muscimol or pentobarbital to the brainstem of a brainstem-spinal cord preparation from neonatal rats decreases the frequency of respiratory activity recorded from the C4 ventral roots. This decrease in frequency is greater in prenatally nicotine-exposed rats compared to control rats. These results suggest that prenatal nicotine exposure potentiates GABAA receptor-mediated inhibition of respiratory rhythm in neonatal rats109. Microinjection of glycine or muscimol into the preBötC of the brainstem-spinal cord preparation from neonatal rats causes abrupt, reversible apnea. Prenatal nicotine exposure prolongs the apnea duration induced by glycine or muscimol suggesting that prenatal nicotine exposure alters development of GABAergic and glycinergic inhibitory transmission in medullary regions involved in central respiratory control, and that neurons in the preBötC are involved110.
Prenatal nicotine exposure reduces the nicotine-induced increase in respiratory frequency recorded from the C4 ventral root in the en bloc brainstem-spinal cord preparation in neonatal rats suggesting that prenatal nicotine exposure diminishes nAChR-mediated excitation in medullary regions involved in the control of breathing frequency, particularly the preBötC111.
In neonatal lambs with prenatal exposure to nicotine, the ventilatory response to hypoxia (10% O2) is similar to controls during wakefulness; however, the ventilatory response to hypoxia during quiet sleep is reduced. Prenatal nicotine exposure also delays arouse from sleep. These results suggest that prenatal nicotine exposure blunts major elements of the cardiorespiratory defense to hypoxia112.
Conclusion
Our understanding regarding neurochemical control of respiration advanced rapidly in last two decades. Genetically engineered knock-out and knock-in mice provide powerful tools for analyzing the molecular composition of native nAChRs and the role of specific subtypes of receptors in regulation of respiration. Development of in vitro models such as the en bloc brainstem-spinal cord and the medullary slice preparations enables us to bridge gaps in our knowledge between molecular and cellular events and respiratory behavior. Recent studies have led to understanding of the role α4* nAChRs in the preBötC and XII nucleus plays in regulation of respiratory frequency and pattern as well as provided insight into the underlying cellular mechanisms of modulation of neurotransmission and neuronal excitability. There are several prevalent neurological disorders related to central control of breathing such as sleep apnea and SIDS where nAChRs may play a role. Nicotine from maternal smoking (in fact, any environmental tobacco smoke) may predispose newborns to SIDS, and obstructive sleep apnea may involve sleep-related cholinergic modulation of upper airway muscle tone. Insight into the molecular and cellular mechanisms of cholinergic regulation of respiration and of the nicotine actions on development of neonatal central control of breathing will provide a pharmacological basis for prevention and treatment of these disorders.
References
Dikshit BB . Action of acetylcholine on the brain and its occurrence therein. J Physiol 1934; 80: 409–21.
Gesell R, Hansen E, Worzniak J . Humoral intermediation of nerve cell activation in the central nervous system. Am J Physiol 1943; 138: 776–91.
Metz B . Correlation between respiratory reflex and acetylcholine content of pons and medulla. Am J Physiol 1962; 202: 80–2.
Wiemer W . On the effect of acetylcholine on respiration. IV. Effect following injection into the cisterna pontis, cisterna cerebellomedullaris, lateral ventricles and cerebrospinal fluid. Pflugers Arch 1963; 276: 568–78.
Brimblecombe RW . Drugs acting on central cholinergic mechanisms and affecting respiration. In: Widdicombe JG editor. International Encyclopedia of Pharmacology and Therapeutics Section 104: respiratory Pharmacology. Oxford: Pergamon Press; 1981. p175–84.
Murakoshi T, Suzue T, Tamai S . A pharmacological study on respiratory rhythm in the isolated brainstem-spinal cord preparation of the newborn rat. Br J Pharmacol 1985; 86: 95–104.
Gillis RA, Walton DP, Quest JA, Namath IJ, Hamosh P, Dretchen KL . Cardiorespiratory effects produced by activation of cholinergic muscarinic receptors on the ventral surface of the medulla. J Pharmacol Exp Ther 1988; 247: 765–73.
Nattie EE, Li A . Ventral medulla sites of muscarinic receptor subtypes involved in cardiorespiratory control. J Appl Physiol 1990; 69: 33–41.
Burton MD, Nouri K, Baichoo S, Samuels-Toyloy N, Kazemi H . Ventilatory output and acetylcholine: perturbations in release and muscarinic receptor activation. J Appl Physiol 1994; 77: 2275–84.
Burton MD, Nouri M, Kazemi H . Acetylcholine and central respiratory control: perturbations of acetylcholine synthesis in the isolated brainstem of the neonatal rat. Brain Res 1995; 670: 39–47.
Haji A, Furuichi S, Takeda R . Effects on iontophoretically applied acetylcholine on membrane potential and synaptic activity of bulbar respiratory neurones in decerebrate cats. Neuropharmacology 1996; 35: 195–203.
Shao XM, Feldman JL . Acetylcholine modulates respiratory pattern: effects mediated by M3-like receptors in preBötzinger complex inspiratory neurons. J Neurophysiol 2000; 83: 1243–52.
Bellingham MC, Funk GD . Cholinergic modulation of respiratory brain-stem neurons and its function in sleep-wake state determination. Clin Exp Pharmacol Physiol 2000; 27: 132–7.
Boudinot E, Champagnat J, Foutz AS . M(1)/M(3) and M(2)/M(4) muscarinic receptor double-knockout mice present distinct respiratory phenotypes. Respir Physiol Neurobiol 2008; 161: 54–61.
Dev NB, Loeschcke HH . A cholinergic mechanism involved in the respiratory chemosensitivity of the medulla oblongata in the cat. Pflugers Arch 1979; 379: 29–36.
Fukuda Y, Loeschcke HH . A cholinergic mechanism involved in the neuronal excitation by H+ in the respiratory chemosensitive structures of the ventral medulla oblongata of rats in vitro. Pflugers Arch 1979; 379: 125–35.
Haxhiu MA, Mitra J, van Lunteren E, Bruce EN, Cherniack NS . Hypoglossal and phrenic responses to cholinergic agents applied to ventral medullary surface. Am J Physiol 1984; 247: R939–44.
Nattie EE, Wood J, Mega A, Goritski W . Rostral ventrolateral medulla muscarinic receptor involvement in central ventilatory chemosensitivity. J Appl Physiol 1989; 66: 1462–70.
Monteau R, Morin D, Hilaire G . Acetylcholine and central chemosensitivity: in vitro study in the newborn rat. Respir Physiol 1990; 81: 241–53.
Burton MD, Johnson DC, Kazemi H . The central respiratory effects of acetylcholine vary with CSF pH. J Auton Nervous System 1997; 62: 27–32.
Boudinot E, Emery MJ, Mouisel E, Chatonnet A, Champagnat J, Escourrou P, et al. Increased ventilation and CO2 chemosensitivity in acetylcholinesterase knockout mice. Respir Physiol Neurobiol 2004; 140: 231–41.
Kinney HC, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, White WF . Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 1995; 269: 1446–50.
Duncan JR, Paterson DS, Kinney HC . The development of nicotinic receptors in the human medulla oblongata: inter-relationship with the serotonergic system. Auton Neurosci 2008; 144: 61–75.
Lydic R, Douglas CL, Baghdoyan HA . Microinjection of neostigmine into the pontine reticular formation of C57BL/6J mouse enhances rapid eye movement sleep and depresses breathing. Sleep 2002; 25: 835–41.
Gilman S, Chervin RD, Koeppe RA, Consens FB, Little R, An H, et al. Obstructive sleep apnea is related to a thalamic cholinergic deficit in MSA. Neurology 2003; 61: 35–9.
Benarroch EE, Schmeichel AM, Parisi JE . Depletion of cholinergic neurons of the medullary arcuate nucleus in multiple system atrophy. Auton Neurosci 2001; 87: 293–9.
Foutz AS, Boudinot E, Morin-Surun MP, Champagnat J, Gonsalves SF, Denavit-Saubie M . Excitability of 'silent' respiratory neurons during sleep-waking states: an iontophoretic study in undrugged chronic cats. Brain Res 1987; 404: 10–20.
Shao XM, Feldman JL . Mechanisms underlying regulation of respiratory pattern by nicotine in preBötzinger complex. J Neurophysiol 2001; 85: 2461–7.
Shao XM, Feldman JL . Pharmacology of nicotinic receptors in preBötzinger complex that mediate modulation of respiratory pattern. J Neurophysiol 2002; 88: 1851–8.
Bellingham MC, Ireland MF . Contribution of cholinergic systems to state-dependent modulation of respiratory control. Respir Physiol Neurobiol 2002; 131: 135–44.
Kubin L, Fenik V . Pontine cholinergic mechanisms and their impact on respiratory regulation. Respir Physiol Neurobiol 2004; 143: 235–49.
Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL . Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science 1991; 254: 726–9.
Gray PA, Rekling JC, Bocchiaro CM, Feldman JL . Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBötzinger complex. Science 1999; 286: 1566–8.
Feldman JL, Del Negro CA . Looking for inspiration: new perspectives on respiratory rhythm. Nat Rev Neurosci 2006; 7: 232–42.
Tan W, Janczewski WA, Yang P, Shao XM, Callaway EM, Feldman JL . Silencing preBotzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat Neurosci 2008; 11: 538–40.
Janczewski WA, Feldman JL . Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol 2006; 570: 407–20.
Ono T, Ishiwata Y, Inaba N, Kuroda T, Nakamura Y . Hypoglossal premotor neurons with rhythmical inspiratory-related activity in the cat: localization and projection to the phrenic nucleus. Exp Brain Res 1994; 98: 1–12.
Chamberlin NL, Eikermann M, Fassbender P, White DP, Malhotra A . Genioglossus premotoneurons and the negative pressure reflex in rats. J Physiol 2007; 579: 515–26.
Volgin DV, Rukhadze I, Kubin L . Hypoglossal premotor neurons of the intermediate medullary reticular region express cholinergic markers. J Appl Physiol 2008; 105: 1576–84.
Woolf NJ, Butcher LL . Cholinergic systems in the rat brain: IV. Descending projections of the pontomesencephalic tegmentum. Brain Res Bull 1989; 23: 519–40.
Woolf NJ . Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol 1991; 37: 475–524.
Rukhadze I, Kubin L . Mesopontine cholinergic projections to the hypoglossal motor nucleus. Neurosci Lett 2007; 413: 121–5.
Ruggiero DA, Giuliano R, Anwar M, Stornetta R, Reis DJ . Anatomical substrates of cholinergic-autonomic regulation in the rat. J Comp Neurol 1990; 292: 1–53.
Jones BE . Immunohistochemical study of choline acetyltransferase-immunoreactive processes and cells innervating the pontomedullary reticular formation in the rat. J Comp Neurol 1990; 295: 485–514.
Schafer MK, Eiden LE, Weihe E . Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. I. Central nervous system. Neuroscience 1998; 84: 331–59.
Lewis PR, Shute CC . The distribution of cholinesterase in cholinergic neurons demonstrated with the electron microscope. J Cell Sci 1966; 1: 381–90.
Connaughton M, Priestley JV, Sofroniew MV, Eckenstein F, Cuello AC . Inputs to motoneurones in the hypoglossal nucleus of the rat: light and electron microscopic immunocytochemistry for choline acetyltransferase, substance P and enkephalins using monoclonal antibodies. Neuroscience 1986; 17: 205–24.
Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, et al. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci 1996; 16: 2179–90.
Arvidsson U, Riedl M, Elde R, Meister B . Vesicular acetylcholine transporter (VAChT) protein: a novel and unique marker for cholinergic neurons in the central and peripheral nervous systems. J Comp Neurol 1997; 378: 454–67.
Roghani A, Shirzadi A, Butcher LL, Edwards RH . Distribution of the vesicular transporter for acetylcholine in the rat central nervous system. Neuroscience 1998; 82: 1195–212.
Misawa H, Nakata K, Matsuura J, Nagao M, Okuda T, Haga T . Distribution of the high-affinity choline transporter in the central nervous system of the rat. Neuroscience 2001; 105: 87–98.
Koizumi H, Wilson CG, Wong S, Yamanishi T, Koshiya N, Smith JC . Functional imaging, spatial reconstruction, and biophysical analysis of a respiratory motor circuit isolated in vitro. J Neurosci 2008; 28: 2353–65.
Houser CR, Crawford GD, Barber RP, Salvaterra PM, Vaughn JE . Organization and morphological characteristics of cholinergic neurons: an immunocytochemical study with a monoclonal antibody to choline acetyltransferase. Brain Res 1983; 266: 97–119.
Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW . The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res 1990; 526: 45–53.
Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 1989; 284: 314–35.
Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW . Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 1993; 13: 596–604.
Dominguez del Toro E, Juiz JM, Peng X, Lindstrom J, Criado M . Immunocytochemical localization of the α7 subunit of the nicotinic acetylcholine receptor in the rat central nervous system. J Comp Neurol 1994; 349: 325–42.
Dehkordi O, Haxhiu MA, Millis RM, Dennis GC, Kc P, Jafri A, et al. Expression of alpha-7 nAChRs on spinal cord-brainstem neurons controlling inspiratory drive to the diaphragm. Respir Physiol Neurobiol 2004; 141: 21–34.
Dehkordi O, Millis RM, Dennis GC, Coleman BR, Johnson SM, Changizi L, et al. Alpha-7 and alpha-4 nicotinic receptor subunit immunoreactivity in genioglossus muscle motoneurons. Respir Physiol Neurobiol 2005; 145: 153–61.
Quitadamo C, Fabbretti E, Lamanauskas N, Nistri A . Activation and desensitization of neuronal nicotinic receptors modulate glutamatergic transmission on neonatal rat hypoglossal motoneurons. Eur J Neurosci 2005; 22: 2723–34.
Herber DL, Severance EG, Cuevas J, Morgan D, Gordon MN . Biochemical and histochemical evidence of nonspecific binding of alpha7nAChR antibodies to mouse brain tissue. J Histochem Cytochem 2004; 52: 1367–76.
Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, et al. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem 2007; 102: 479–92.
Salmoiraghi GC, Steiner FA . Acetylcholine sensitivity of cat's medullary neurons. J Neurophysiol 1963; 26: 581–97.
Jordan D, Spyer KM . Effects of acetylcholine on respiratory neurones in the nucleus ambiguus-retroambigualis complex of the cat. J Physiol 1981; 320: 103–11.
Bradley PB, Lucy AP . Cholinoceptive properties of respiratory neurones in the rat medulla. Neuropharmacology 1983; 22: 853–58.
Böhmer G, Schmid K, Schmidt P, Stehle J . Cholinergic effects on spike-density and burst-duration of medullary respiration-related neurones in the rabbit: an iontophoretic study. Neuropharmacology 1987; 26: 1561–72.
Böhmer G, Schmid K, Baumann M . Evidence for a respiration-modulated cholinergic action on the activity of medullary respiration-related neurons in the rabbit. An iontophoretic study. Pflugers Arch 1989; 415: 72–80.
Foutz AS, Boudinot E, Denavit-Saubie M . Central respiratory depression induced by acetylcholinesterase inhibition: involvement of anaesthesia. Eur J Pharmacol 1987; 142: 207–13.
Stewart WC, Anderson EA . Effect of a cholinesterase inhibitor when injected into the medulla of the rabbit. J Pharmacol Exp Ther 1968; 162: 309–18.
Connelly CA, Dobbins EG, Feldman JL . Pre-Bötzinger complex in cats: respiratory neuronal discharge patterns. Brain Res 1992; 590: 337–40.
Schwarzacher SW, Smith JC, Richter DW . Pre-Bötzinger complex in the cat. J Neurophysiol 1995; 73: 1452–61.
Mutolo D, Bongianni F, Carfi M, Pantaleo T . Respiratory changes induced by kainic acid lesions in rostral ventral respiratory group of rabbits. Am J Physiol Regul Integr Comp Physiol 2002; 283: R227–42.
Mutolo D, Bongianni F, Nardone F, Pantaleo T . Respiratory responses evoked by blockades of ionotropic glutamate receptors within the Botzinger complex and the pre-Bötzinger complex of the rabbit. Eur J Neurosci 2005; 21: 122–34.
Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED . Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend 1993; 33: 23–9.
Butera R, Rinzel J, Smith J . Models of respiratory rhythm generation in the pre-Bötzinger complex. II. Populations of coupled pacemaker neurons. J Neurophysiol 1999; 82: 398–451.
Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K . Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci 1999; 20: 142–50.
Bencherif M, Lovette ME, Fowler KW, Arrington S, Reeves L, Caldwell WS, et al. RJR-2403: a nicotinic agonist with CNS selectivity I. In vitro characterization. J Pharmacol Exp Ther 1996; 279: 1413–21.
Hatori E, Sakuraba S, Kashiwagi M, Kuribayashi J, Tsujita M, Hosokawa Y, et al. Association of nicotinic acetylcholine receptors with central respiratory control in isolated brainstem-spinal cord preparation of neonatal rats. Biol Res 2006; 39: 321–30.
Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 2004; 306: 1029–32.
Shao XM, Tan W, Xiu J, Puskar N, Fonck C, Lester HA, et al. α4* nicotinic receptors in preBötzinger complex mediate cholinergic/nicotinic modulation of respiratory rhythm. J Neurosci 2008; 28: 519–28.
Shao XM, Feldman JL . Efficient measurement of endogenous neurotransmitters in small localized regions of central nervous systems in vitro with HPLC. J Neurosci Methods 2007; 160: 256–63.
Shao XM, Feldman JL . Cholinergic neurotransmission in the preBötzinger complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience 2005; 130: 1069–81.
Greer JJ, Smith JC, Feldman JL . Role of excitatory amino acids in the generation and transmission of respiratory drive in neonatal rat. J Physiol 1991; 437: 727–49.
Funk GD, Smith JC, Feldman JL . Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J Neurophysiol 1993; 70: 1497–515.
Aldes LD . Subcompartmental organization of the ventral (protrusor) compartment in the hypoglossal nucleus of the rat. J Comp Neurol 1995; 353: 89–108.
Dobbins EG, Feldman JL . Differential innervation of protruder and retractor muscles of the tongue in rat. J Comp Neurol 1995; 357: 376–94.
Fuller DD, Williams JS, Janssen PL, Fregosi RF . Effect of co-activation of tongue protrudor and retractor muscles on tongue movements and pharyngeal airflow mechanics in the rat. J Physiol 1999; 519: 601–13.
Zaninetti M, Tribollet E, Bertrand D, Raggenbass M . Presence of functional neuronal nicotinic acetylcholine receptors in brainstem motoneurons of the rat. Eur J Neurosci 1999; 11: 2737–48.
Chamberlin NL, Bocchiaro CM, Greene RW, Feldman JL . Nicotinic excitation of rat hypoglossal motoneurons. Neuroscience 2002; 115: 861–70.
Robinson DM, Peebles KC, Kwok H, Adams BM, Clarke LL, Woollard GA, et al. Prenatal nicotine exposure increases apnoea and reduces nicotinic potentiation of hypoglossal inspiratory output in mice. J Physiol 2002; 538: 957–73.
Liu X, Sood S, Liu H, Horner RL . Opposing muscarinic and nicotinic modulation of hypoglossal motor output to genioglossus muscle in rats in vivo. J Physiol 2005; 565: 965–80.
Gothe B, Strohl KP, Levin S, Cherniack NS . Nicotine: a different approach to treatment of obstructive sleep apnea. Chest 1985; 87: 11–17.
Hedner J, Kraiczi H, Peker Y, Murphy P . Reduction of sleep-disordered breathing after physostigmine. Am J Respir Crit Care Med 2003; 168: 1246–51.
Zhang RX, Hui N . Effect of intrathecal injection of acetylcholine on phrenic nerve firing activity in rabbits. Sheng Li Xue Bao 1991; 43: 89–93. Chinese.
Chitravanshi VC, Sapru HN . Microinjections of carbachol into the phrenic motor nucleus inhibit phrenic nerve activity in the rat. Brain Res 1999; 837: 298–300.
Mathews TJ, MacDorman MF . Infant mortality statistics from the 2004 period linked birth/infant death data set. Natl Vital Stat Rep 2007; 55: 1–32.
Moon RY, Horne RS, Hauck FR . Sudden infant death syndrome. Lancet 2007; 370: 1578–87.
Thach BT . The role of respiratory control disorders in SIDS. Respir Physiol Neurobiol 2005; 149: 343–53.
Blair PS, Sidebotham P, Berry PJ, Evans M, Fleming PJ . Major epidemiological changes in sudden infant death syndrome: a 20-year population-based study in the UK. Lancet 2006; 367: 314–9.
Mitchell EA, Milerad J . Smoking and the sudden infant death syndrome. Rev Environ Health 2006; 21: 81–103.
Nachmanoff DB, Panigrahy A, Filiano JJ, Mandell F, Sleeper LA, Valdes-Dapena M, et al. Brainstem 3H-nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol 1998; 57: 1018–25.
Slotkin TA, Orband-Miller L, Queen KL . Development of 3H]nicotine binding sites in brain regions of rats exposed to nicotine prenatally via maternal injections or infusions. J Pharmacol Exp Ther 1987; 242: 232–7.
Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA . Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull 1989; 23: 187–92.
Frank MG, Srere H, Ledezma C, O'Hara B, Heller HC . Prenatal nicotine alters vigilance states and AChR gene expression in the neonatal rat: implications for SIDS. Am J Physiol Regul Integr Comp Physiol 2001; 280: R1134–40.
Pentel PR, Keyler DE, Chen Y, LeSage MG, Dufek MB, Le C, et al. Vaccination against nicotine does not prevent nicotine-induced changes in fetal nicotinic receptor binding and c-fos mRNA expression in rats. Neurotoxicol Teratol 2006; 28: 589–96.
Fewell JE, Smith FG, Ng VK . Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir Physiol 2001; 127: 61–73.
Huang YH, Brown AR, Costy-Bennett S, Luo Z, Fregosi RF . Influence of prenatal nicotine exposure on postnatal development of breathing pattern. Respir Physiol Neurobiol 2004; 143: 1–8.
Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H . Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci USA 2005; 102: 3817–21.
Luo Z, Costy-Bennett S, Fregosi RF . Prenatal nicotine exposure increases the strength of GABA(A) receptor-mediated inhibition of respiratory rhythm in neonatal rats. J Physiol 2004; 561: 387–93.
Luo Z, McMullen NT, Costy-Bennett S, Fregosi RF . Prenatal nicotine exposure alters glycinergic and GABAergic control of respiratory frequency in the neonatal rat brainstem-spinal cord preparation. Respir Physiol Neurobiol 2007; 157: 226–34.
Fregosi RF, Pilarski JQ . Prenatal nicotine exposure and development of nicotinic and fast amino acid-mediated neurotransmission in the control of breathing. Respir Physiol Neurobiol 2008; 164: 80–6.
Hafstrom O, Milerad J, Sundell HW . Prenatal nicotine exposure blunts the cardiorespiratory response to hypoxia in lambs. Am J Respir Crit Care Med 2002; 166: 1544–9.
Acknowledgements
This work was supported by Tobacco-Related Disease Research Program (California) grant 13QT-0164 and NIH Grant HL40959.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shao, X., Feldman, J. Central cholinergic regulation of respiration: nicotinic receptors. Acta Pharmacol Sin 30, 761–770 (2009). https://doi.org/10.1038/aps.2009.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.88
Keywords
This article is cited by
-
The lesion site of organophosphorus-induced central apnea and the effects of antidotes
Scientific Reports (2023)
-
Basal forebrain cholinergic signalling: development, connectivity and roles in cognition
Nature Reviews Neuroscience (2023)
-
Silencing A7-nAChR levels increases the sensitivity of gastric cancer cells to ixabepilone treatment
Tumor Biology (2016)
-
Can donepezil facilitate weaning from mechanical ventilation in difficult to wean patients? An interventional pilot study
DARU Journal of Pharmaceutical Sciences (2015)
-
Incidence and determinants of sudden infant death syndrome: a population-based study on 37 million births
World Journal of Pediatrics (2015)
