
Abstract
Currently, cardiovascular diseases remain one of the leading causes of morbidity and mortality in the world, indicating the need for innovative therapies and diagnosis for heart disease. MicroRNAs (miRNAs) have recently emerged as one of the central players in regulating gene expression. Numerous studies have documented the implications of miRNAs in nearly every pathological process of the cardiovascular system, including cardiac arrhythmia, cardiac hypertrophy, heart failure, cardiac fibrosis, cardiac ischemia and vascular atherosclerosis. More surprisingly, forced expression or suppression of a single miRNA is enough to cause or alleviate the pathological alteration, underscoring the therapeutic potential of miRNAs in cardiovascular diseases. In this review we summarize the key miRNAs that can solely modulate the cardiovascular pathological process and discuss the mechanisms by which they exert their function and the perspective of these miRNAs as novel therapeutic targets and/or diagnostic markers. In addition, current approaches for manipulating the action of miRNAs will be introduced.
Similar content being viewed by others

Introduction
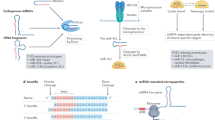
MicroRNAs (miRNAs) are endogenous, conserved ∼22-nucleotide non-coding RNAs that anneal to inexactly complementary sequences in the 3′-untranslated regions (3′UTR) of target mRNAs of protein-coding genes to cause mRNA cleavage or repression of the translational machinery for protein synthesis1. The first miRNA lin-4 was discovered in 1993, which regulates C elegans development by inhibiting the protein expression of lin-14 via binding to the 3′UTR of its mRNA2. In 2000, the second miRNA let-7 was identified as a 21-nucleotide small RNA that has a complementary sequence to the 3′UTR of lin-14 and participates in the regulation of C elegans development3. After that, a large number of miRNAs have been found and a series of research uncovered the functional role of miRNAs in diverse biological and pathophysiological processes4, 5. Among them, the involvement of miRNAs in the pathogenesis of cardiovascular diseases has recently been intensively investigated6, 7, 8.
MiRNAs are initially transcribed as long RNA precursors called primary miRNAs that require the RNase III enzyme Drosha in the nucleus to trim them into precursor miRNAs. The latter, characterized by a stem loop or hairpin structure of 70–100 nt, is exported by the nuclear export factor exportin-5 to the cytoplasm where they are subsequently cropped to become mature miRNAs of 21–26 nt long by another RNase III enzyme Dicer9, 10, 11, 12, 13. Mature miRNAs can interact with Argonaute to form the RNA-induced silencing complex (RISC) and then guide the RISC to their target mRNAs, most favorably to the 3′UTR. In order for a miRNA to produce functional consequences, its 5′-end 2 to 8 nts must have exact base pairing to the target mRNA, the “seed” region, and partial complementarity with the rest of the sequence. An miRNA can either inhibit translation or induce degradation of its target mRNA, primarily depending upon the overall degree of complementarity of the binding site, number of binding sites, and the accessibility of the bindings sites (determined by free energy states). The greater the complementarity of the accessible binding sites, the more likely an miRNA degrades its targeted mRNA, and those miRNAs that display imperfect sequence complementarities with target mRNAs primarily result in translational inhibition14, 15, 16. MiRNAs are an abundant RNA species constituting >2% of the predicted human genes (>1000 genes), which regulates ∼30% of protein–coding genes. Some miRNAs are expressed at >1000 copies per cell17.
With the recent surge of research into miRNAs, this category of endogenous non-coding small ribonucleic acids has emerged as one of the central players of gene expression regulation. The research on miRNAs in relation to cardiovascular disease has become a most rapidly evolving field. The dysregulation of many individual miRNAs has been linked to the development and progression of cardiovascular disease. Forced expression or suppression of a single miRNA is enough to cause or alleviate the pathological change. The role of miRNAs in the pathogenesis of the heart and vessels points to a possibility of miRNAs as targets for treatment of cardiovascular disease. In this review, the aberrantly expressed miRNAs that underlie the development of cardiovascular diseases will be discussed. The therapeutic potential of miRNAs as new targets for cardiovascular diseases and strategies for manipulating miRNAs by influencing their expression, stability, and function will be introduced.
Therapeutic implications of miRNAs in cardiovascular diseases
miRNA and cardiac hypertrophy and heart failure
In response to stress (such as hemodynamic alterations associated with myocardial infarction, hypertension, aortic stenosis, valvular dysfunction, etc), the adult heart undergoes remodeling process and hypertrophic growth to adapt to altered workloads and to compensate for the impaired cardiac function. 'The remodeled heart frequently progresses into overt heart failure. Hypertrophic growth manifests enlargement of cardiomyocyte size and enhancement of protein synthesis through the activation of intracellular signaling pathways and transcriptional mediators in cardiac myocytes. The process is characterized by a reprogramming of cardiac gene expression and the activation of “fetal” cardiac genes18. In light of the role of miRNA in gene regulation, miRNAs may be involved in cardiac hypertrophy and heart failure. This concept was confirmed by a number of reports which demonstrated that dysregulation of a single miRNA is sufficient to induce this pathological alteration, pointing to miRNAs as potential novel therapeutic targets in cardiac hypertrophy and heart failure.
To date, 11 miRNAs have been experimentally established to determine this pathological process. These include miR-1, 133, 129, 18b, 195, 21, 23a, 23b, 24, 208, and 212. Of them, miR-1 and miR-133 are anti-hypertrophic, and miR-129, 18b, 195, 23a, 23b, 24, 208, and 212 are pro-hypertrophic. However, the role of miR-21 in cardiac hypertrophy is still controversial19, 20, 21, 22, 23, 24, 25, 26, 27, 28. The first group of miRNAs regulating cardiac hypertrophy were identified in Olson's lab20. They described a typical signature pattern of miRNAs in cardiac tissue from mice in response to transverse aortic constriction (TAC) or expression of activated calcineurin, stimuli that induce pathological cardiac remodeling hypertrophic. They further showed that forced overexpression of individual stress-inducible miRNAs eg. miR-195, 23a, 23b, 24, or miR-195 alone induced hypertrophy in cultured cardiomyocytes. More interestingly, transgenic overexpression of miR-195 in mice was sufficient to induce pathological cardiac growth and heart failure as indicated by thinning of the left ventricular walls, increase in left ventricular diameter and deterioration in cardiac function. Olson's group then investigated the effect of miR-20819, a cardiac-specific miRNA that is encoded by an intron of the α-MHC gene, on cardiac hypertrophy. They found that miR-208 is required for cardiomyocyte hypertrophy, fibrosis, and expression of β-MHC in response to stress and hypothyroidism. Mice homozygous for miR-208 deletion failed to undergo stress-induced cardiac remodeling, hypertrophic growth, and β-MHC upregulation, whereas transgenic expression of miR-208 was sufficient to induce β-MHC and hypertrophy. The thyroid hormone receptor associated protein 1 (THRAP1), a cofactor of the thyroid hormone receptor, was verified to be the target mRNA of miR-208. By repressing THRAP1, miR-208 regulates the expression of β-MHC and, thereby, the development of hypertrophy to stress. A recent study by Wang DZ et al confirmed the action of miR-208 by discovering that transgenic overexpression of miR-208a in the heart induced hypertrophic growth in mice, which resulted in pronounced repression of the miR-208 regulatory targets thyroid hormone–associated protein 1 and myostatin, 2 negative regulators of muscle growth and hypertrophy29.
Sayed et al24 explored the role of miR-1, a muscle-specific miRNA, in pressure-overload induced cardiac hypertrophy. They found that miR-1 was singularly downregulated as early as day 1, persisting through day 7, after TAC-induced hypertrophy in a mouse model. Moreover, the aberrant expressed miR-1 is a causative factor of hypertrophy. Overexpression of miR-1 carried by adenovirus vector prevented hypertrophic growth of neonatal cardiac myocytes by inhibiting its growth-related targets, including Ras guanosine-triphosphatase-activating protein, cyclin-dependent kinase 9, fibronectin, and Ras homolog enriched in brain. Consistently, Caré et al28 confirmed the down-regulation of miR-1 in three different hypertrophic models, including transverse aortic arch–constricted mice, transgenic mice with selective cardiac overexpression of a constitutively active mutant of the Akt kinase, and exercised rats, and the inhibitory role of miR-1 on cardiac hypertrophy in vitro. miR-1 was also reported to negatively regulate the expression of hypertrophy-associated genes, Mef2a and Gata4, and attenuate cardiomyocyte hypertrophy in cultured neonatal rat cardiomyocytes and in the intact adult heart25.
The antihypertrophic action of another muscle-specific miRNA, miR-133, was described in two studies. Caré et al28 found that the expression of miR-133, which transcribed together with miR-1 as a bicistronic cluster, was decreased in the left ventricle of the above mentioned three hypertrophic models. In vitro overexpression of miR-133 inhibited cardiac hypertrophy. In contrast, suppression of miR-133 induced hypertrophy, which was more pronounced than that after stimulation with conventional inducers of hypertrophy. In vivo inhibition of miR-133 by a single infusion of a chemically modified antisense RNA oligonucleotide (termed 'antagomiR') targeted to miR-133 lead to one month miR-133 knockdown and caused marked and sustained cardiac hypertrophy. To elucidate the molecular mechanism underlying miR-133's control on cardiac hypertrophy, they identified specific targets of miR-133: RhoA, a guanosine diphosphate–guanosine triphosphate exchange protein regulating cardiac hypertrophy; Cdc42, a signal transduction kinase implicated in hypertrophy; and Nelf-A/WHSC2, a nuclear factor involved in cardiogenesis. Li PF's group found that miR-23a is a pro-hypertrophic miRNA, which is regulated by the transcription factor, nuclear factor of activated T cells (NFATc3). Under hypertrophic stimuli the expression of miR-23a was up-regulated, which leads to cardiac hypertrophy via targeting the muscle specific ring finger protein 1, an anti-hypertrophic protein. More excitingly, they demonstrated that knockdown of miR-23a using antagomir technique could significantly attenuate hypertrophy induced by isopropranolol, suggesting the potential of miR-23a as an anti-hypertrophic target30. Sucharov et al demonstrated that over-expression of miR-133b prevents changes in gene expression patterns mediated by β-adrenergic receptor stimulation23.
Simultaneous overexpression of three fetal miRNAs up-regulated in the failing heart (miR-21, miR-129, and miR-212) resulted in hypertrophic morphological changes of neonatal cardiomyocytes similar to that observed in the failing heart. Meanwhile, a number of both established (ANP, BNP, β-MHC, α-skeletal actin) and newly identified fetal genes (villin2, cspg2, phlda1, hsp90, RASA1, MEF2a, cradd, dtna) were reactivated or silenced by this modulation of the miRNA environment. Interestingly, overexpression of a single miRNA (miR-21, miR-129, or miR-212) had only minor effects, indicating that certain pathological changes may require an alteration of the miRNA environment21. Another study also confirmed the upregulation of miR-21 in hypertrophic heart and in cultured neonatal hypertrophic cardiomyocytes stimulated by angiotensin II or phenylephrine, and the antihypertrophic action of miR-21 was obtained by suppressing its aberrant expression via antisense-mediated depletion27. However, a contradictory result was also reported22, namely that miR-21 produced antihypertrophic rather than prohypertrophic action.
miRNA and arrhythmia
Arrhythmias are electrical disturbances that can result in irregular heart beating, which is the leading cause of sudden cardiac death. Alterations of the three intrinsic properties of the heart, including impaired excitation conduction/propagation, enhanced automaticity, or abnormal repolarization lead to disturbed excitability, manifesting as arrhythmias. Cardiac ion channels are fundamental determinants of cardiac intrinsic properties. Malfunction of these ion channels either caused by mutations in the genes encoding the channel proteins or altered expression under proarrhythmic conditions, such as cardiac remodeling, ischemia etc, can predispose to arrhythmias. Conventional pharmacologic agents targeting ion channels remain the major therapeutic tools in treating cardiac arrhythmias. However, the limited efficacy and proarrhythmic potential of antiarrhythmic agents restricts their application. Recent findings that miRNAs regulate expression of cardiac ion channels strongly indicate a role of these miRNAs to influence arrhythmogenicity and a potential of these miRNAs being the new antiarrhythmic targets31. Life-threatening ischemic arrhythmias are common clinical complications in ischemic heart diseases. Our group found that the expression level of miR-1 in the myocardium of coronary artery disease patients was significantly increased. In vivo application of miR-1 into the myocardium induces arrhythmias in both healthy normal hearts and a rat model of myocardial infarction31. Knock down of miR-1 using its specific inhibitor antisense oligonucleotides (AMO) reversed these effects. GJA1/Cx43 and KCNJ2/Kir2.1 are the two targets of miR-1, which explains the arrhythmogenic action of miR-1, as decreased expression of Cx43 and Kir2.1 during cardiac ischemia underlies the occurrence of ischemic arrhythmia. These data indicated that myocardial infarction upregulates miR-1 expression via some unknown factors, which induces post-transcriptional repression of GJA1 and KCNJ2, resulting in conduction slowing that finally leading to ischemic arrhythmias31. This work was regarded as an exciting step in the dissection of new molecular signaling pathways for arrhythmias and sudden death32. Dmitry Terentyev et al33 further confirmed the arrhythmogenic action of miR-1. They found that in the presence of isoproterenol, rhythmically paced, miR-1-overexpressing myocytes exhibited spontaneous arrhythmogenic oscillations of intracellular Ca2+, events that occurred rarely in control myocytes under the same conditions. Protein phosphatase PP2A was validated as a new target of miR-1. Suppression of PP2A by miR-1 selectively leads to CaMKII-dependent hyperphosphorylation of RyR2, enhances RyR2 activity, and promotes arrhythmogenic SR Ca2+ release.
Atrial fibrillation is another most commonly encountered cardiac arrhythmia and is directly or indirectly responsible for considerable mortality and morbidity in cardiovascular disease patients. Elucidation of the molecular mechanisms underlying the development of atrial fibrillation may provide novel targets for future therapy. Our group found that miR-133 and miR-590 are involved in atrial structural remodeling, which is the major pathological basis for the development of atrial fibrillation34. In the atrium of nicotine treated dogs, TGF-β1 and TGF-β RII were upregulated at the protein level, while miR-133 and miR-590 were downregulated. TGF-β1 and TGF-βRII were verified as the targets of miR-133 and miR-590, respectively, as transfection of miR-133 or miR-590 into cultured atrial fibroblasts decreased levels of TGF-β1 and TGF-βRII and collagen content, which were abolished by the antisense oligonucleotides against miR-133 or miR-59034. This study indicates that interference with miRNAs using small silencing RNAs or antisense oligonucleotides has the potential to be developed as novel therapeutic approach to AF35. Interestingly, the cardiac specific miRNA miR-208a was demonstrated to participate in both cardiac conduction abnormality and the occurance of AF, as miR-208a transgenic mice manifested first-degree atrial-ventricular block, while miR-208a knockout mice developed AF, which strongly indicates the importance of maintaining the physiological balance of miRNA levels29.
Pacemaker channels, which carry the nonselective cation currents, are critical in generating sinus rhythm and ectopic heart beats under various pathological settings such as cardiac hypertrophy. Luo et al showed that miR-1 and miR-133 also targeted pacemaker channels, HCN2 and HCN4, with HCN2 modulated by miR-1 and miR-133 and HCN4 by miR-136. Forced expression of miR-1/miR-133 by transfection prevented overexpression of HCN2/HCN4 in hypertrophic cardiomyocytes, indicating its antiarrhythmic capability. Rapid delayed rectifier K+ current (IKr), encoded by human ether-á-go-go-related gene (HERG), and slow delayed rectifier K+ current (IKs), encoded by KCNQ1 assembles with KCNE1, are two potassium currents responsible for the repolarization of cardiac myocytes and play a critical role in governing cardiac APD. Impairment of these repolarization current channels can cause substantial prolongation of APD or QT interval prolongation, which is associated with an increased risk of sudden cardiac death consequent to lethal ventricular arrhythmias. Xiao et al tested the involvement of miRNAs underlying the discrepancy expression of ERG channels in a rabbit diabetic model. They found that repression of ERG by miR-133 in diabetic hearts likely underlies the differential changes of ERG protein and transcript, thereby causing depression of IKr and repolarization slowing, resulting in QT prolongation and arrhythmias37. In addition, they experimentally established that KCNQ1 and KCNE1 are targets of miR-133 and miR-1, respectively38. More importantly, they found that the distribution of miR-133 and miR-1 transcripts within the heart is also spatially heterogeneous with the patterns corresponding to the spatial distribution of KCNQ1 and KCNE1 proteins and IKs. Zhao et al39 found that targeted deletion of the muscle-specific miRNA, miR-1-2, lead to abnormal propagation of cardiac electrical activity, manifested as slower heart rate, a shortened PR interval, and a broadened QRS complex, indicative of bundle branch block. They further validated the miR-1-2 targets, including the cardiac transcription factor, Irx5, which represses KCND2, a potassium channel subunit (Kv4.2) responsible for transient outward K+ current (Ito). Clearly, the multi-target effects of muscle-specific miR-1 and miR-133 on ion channels supports a central role of miRNA for fine tuning the regulation of cardiac electrophysiology and the potential of them as novel therapeutic targets.
miRNA and cardiac fibrosis
Cardiac fibrosis, a process of excessive extracellular matrix (ECM) protein accumulation, also called ECM remodeling, is an important feature of cardiac adaptation to numerous pathological stimuli, such as hypertension and cardiac infarction. This process alters mechanical stiffness and electric properties of the heart and contributes to diastolic dysfunction, cardiomyocyte loss, arrhythmias, and the progression to heart failure. Alleviation of ECM remodeling was considered a reasonable strategy to improve cardiac function and prevent heart failure associated with cardiac infarction and hypertension. Interestingly, several miRNAs including miR-133, miR-21, miR-29 and miR-30 have been demonstrated to participate in the regulation of ECM remodeling. Olson's group40 found that the miR-29 family of miRNAs, which consists of three members expressed from two bicistronic miRNA clusters (miR-29b-1 is co-expressed with miR-29a, and miR-29b-2 is co-expressed with miR-29c), are downregulated in the region of the heart adjacent to the infarct in chronic cardiac infarction rats. In vivo knockdown of cardiac miR-29b by tail vein injection of cholesterol-modified oligonucleotides (antagomiR miR-29b) modestly increased collagen expression in the heart, whereas over-expression of miR-29 in fibroblasts reduced collagen expression, indicating the potential role of miR-29 as a negative regulator of collagen expression. A large number of mRNAs that encode proteins involved in fibrosis, including multiple collagens, fibrillins, and elastin were found to be targets of miR-29.
The role of miR-21 in the process of fibrosis has been investigated in-depth41, 42. miR-21 was reported to be progressively upregulated in cardiac fibroblasts during cardiac ischemia/reperfusion42 or the later stages of heart failure41. Upregulation of miR-21 in cardiac fibroblasts results in inhibition of SPRY1 protein expression, augmentation of ERK–MAP kinase activity, which in turn enhances cardiac fibroblast survival and thereby the interstitial fibrosis and cardiac remodeling that is characteristic of the failing heart41. More importantly, blocking of endogenous miR-21 by antagomiR-21, which is a chemically modified antisense oligonucleotide specific for miR-21, in pressure overload mice reduced genes encoding collagens and extracellular matrix molecules that are highly upregulated during cardiac fibrosis, and significantly attenuated the increased interstitial fibrosis, cardiomyocyte size and heart weight. This finding may provide a new remedy for the treatment of fibrosis.
Different from miR-29 and miR-21, another two miRNAs, miR-133 and miR-30c, are down regulated during ventricular hypertrophy43. Overexpression of miR-133 or miR-30c in cultured fibroblasts decreased the production of collagens by suppressing connective tissue growth factor (CTGF), which is a powerful inducer of ECM synthesis and contributes to myocardial remodeling43.
miRNA and cardiac ischemic disease
Ischemic/reperfusion injury is one of the most common cardiovascular diseases that requires intensive research to fully elucidate the underlying mechanisms and improve the therapeutic outcomes. Oxidative stress and apoptosis are the key pathological processes that medicate ischemic/reperfusion injury. Induction of neovascularization and ischemic preconditioning has been established as effective cardioprotective strategies. Increasing evidence indicates that miRNAs, such as miR-1/106, miR-133, miR-21, miR-320, miR-199a and miR-92a, are also implicated in ischemic cardiac disease. We have recently shown that miR-1 level is markedly elevated in ischemic myocardium where apoptotic cell death plays an important role in the detrimental changes of the diseased heart31. Coincidently, another study from our laboratories revealed a novel aspect of cellular functions of the muscle-specific miRNAs miR-1 and miR-133, ie regulation of apoptosis and survival in cardiomyocytes. A unique feature of this regulation is the opposing actions with miR-1 being pro-apoptotic and miR-133 being anti-apoptotic, suggesting a possible role of relative miR-1 and miR-133 levels in regulating the cell fate. Post-transcriptional repression of heat shock protein 60 and heat shock protein 70 by miR-1 and of caspase 9 by miR-133 is probably one of the mechanisms underlying their regulation of apoptosis versus survival44. Our results therefore suggest that the ischemic elevation of miR-1 level may contribute to the enhanced apoptotic cell death in myocardial infarction, in addition to the enhanced arrhythmogenicity. This conception was proven by two studies from other researchers. Yu et al45 reported that miR-1 is a key mediator of glucose induced apoptosis46 and suppression of insulin-like growth factor-1 expression represents a new mechanism underlying its proapoptotic action. Furthermore, we found that miR-1 also participates in the regulation of beta-blocker produced cardioprotection47. Administration of propranolol reversed the up-regulation of miR-1 nearly back to the control level during ischemia, which then lead to relieving of myocardial injury by restoring the membrane depolarization and cardiac conduction slowing, and rescuing the expression of inward rectifying K+ channel subunit Kir2.1 and gap junction channel connexin 43 (Figure 1).

Summary of beta-adrenergic blocker produced protective effects against ischemic arrhythmias and cardiac injury47. Beta-blockers reduced miR-1 level via inhibiting beta-adrenoceptor-cAMP-PKA pathway and a transcriptional factor-serum response factor (SRF), which led to up-regulated expression of Cx43 formed gap junctions and inward rectifier potassium channels (IK1).
Another miRNA, miR-21, was found to prevent cell damage subjected to H2O2, which mimics the pathological condition of high levels of reactive oxygen species (ROS) that act as an important inducer of cardiac cell injury48. Consistently, another study proved that miR-21 demonstrated a significant increase in mice heart subjected to cytoprotective heat-shock (HS). Moreover, overexpression of chemically synthesized miR-21 reduced infarct size which was associated with the inhibition of pro-apoptotic genes and increase in anti-apoptotic genes, whereas a miR-21 inhibitor abolished this effect49. In a mouse model of myocardial infarction, systemic administration of an antagomiR designed to inhibit miR-92a resulted in enhanced blood vessel growth and functional recovery of damaged tissue50. miR-92a appears to target mRNAs corresponding to several pro-angiogenic proteins, including the integrin subunit alpha550. Ren et al51 demonstrated that miR-320 is involved in the regulation of ischemia/reperfusion (I/R)-induced cardiac injury and dysfunction via antithetical regulation of Hsp20. Transgenic overexpression of miR-320 increased apoptosis and infarction size in the hearts on I/R, whereas in vivo treatment with antagomir-320 reduced infarction size relative to the administration of mutant antagomir-320 and saline controls. These studies not only elucidated the complex molecular mechanisms of cardiac injury in a new level, but also presented valuable therapeutic targets in the setting of ischemic disease.
miRNA and vascular disease
Aberrant proliferation of vascular smooth muscle cells (VSMCs) and the formation of neointimal lesion is a key pathological process of a variety of proliferative vascular diseases, such as atherosclerosis, coronary heart diseases, postangioplasty restenosis and transplantation arteriopathy52. Recently, Zhang's group demonstrated the regulative role of miRNAs including miR-21, miR-221, and miR-222 in the process of aberrant VSMCs proliferation53, 54. They found that miR-21, miR-221, and miR-222 were significantly upregulated in carotid arteries after angioplasty. Depletion of the aberrantly overexpressed miR-21, miR-221, or miR-222, via antisense-mediated knockdown, has a significantly negative effect on neointimal lesion formation in rat artery after angioplasty, while forced overexpression of these miRNAs promotes VSMCs proliferation. They further confirmed that PTEN and Bcl-2, two important signal molecules associated with VSMC growth and apoptosis, are the targets of miR-21, through which miR-21 exerts its function53. They then elucidated that p27(Kip1) and p57(Kip2) are related to miR-221– and miR-222–mediated effect on VSMC proliferation, as in the VSMCs deficient of these genes the inhibitory effect of miR-221 and miR-222 on VSMC proliferation is decreased54. These results indicate that miRNAs are important regulators in the development of proliferative vascular diseases and the potential therapeutic targets in vascular diseases.
MiRNAs based therapeutic strategies
The recent studies on the role of miRNAs in cardiovascular diseases imply: (1) miRNAs are key regulators of a variety of cardiovascular diseases, including hypertrophy, heart failure, cardiac injury, arrhythmia and asthersclerosis etc; (2) Down- or up-regulation of one particular miRNA is enough to cause the pathogenesis of specific cardiovascular disease, which can be reversed by correcting the aberrant expressed miRNA, indicating the potential of miRNAs as pharmacological targets. Therefore, approaches to manipulate either positively or negatively the expression of miRNAs can be taken for therapeutic purposes. For those miRNAs that are downregulated under disease states, forced miRNA re-expression strategy is used to recover miRNA expression, whereas for those miRNAs upregulated, anti-miRNA strategy is employed to suppress their expression.
miRNA re-expression strategy
To recover the decreased miRNA levels in cardiovascular diseases and thus prevent the pathological process, the exogenous miRNAs either synthesized artificially or constructed in virus vectors have been often used in previous studies to achieve optimistic outcomes24, 41. Generally, these miRNAs are double-stranded and have the same sequence as endogenous miRNAs.
However, as one miRNA can target hundreds of mRNAs, the inhibitory action of a particular miRNA can be nonspecific and the off-target effect by an exogenous miRNA seems inevitable. In order to circumvent the problem of non-gene specificity of miRNA actions, a technology named miRNA Mimic technology was developed, which utilizes synthetic, non-natural nucleic acids that can bind to the unique sequence of target mRNAs in a gene-specific manner and elicits post-transcriptional regulatory effects as a miRNA does55. A miRNA mimic must be complementary to, only to, the 3′UTR of the target gene to elicit miRNA action, that is repressing the target gene at the post-transcriptional level with minimal effects on the mRNA level. Meanwhile, the 3′UTR of the target gene must contain a unique sequence distinct from other genes to elicit gene-specific action. This technique was successfully verified by applying it to cardiac pacemaker genes HCN2 and HCN455. After identifying a stretch of sequence in the 3′UTR unique to the HCN2 (or HCN4) gene that is expectedly long enough for miRNA action, a 22-nt miRNA mimic complementary to the HCN2 (or HCN4) sequence was designed, which has eight nucleotides (nucleotides 2–8) at the 5′ end, and seven nucleotides at the 3'end. These miRNA mimics substantially repressed HCN channel protein expression, with concomitant depression of pacemaker activities and reduction of heart rate but with minimal effects on their mRNA levels. The results demonstrated a promise of utilizing the technology for gene-specific repression of expression at the protein level based on the principle of miRNA actions.
Anti-miRNA strategy
For those miRNAs that are aberrantly upregulated and play a causal role in disease states, the anti-miRNA techniques that suppress their expression should be employed. Inhibiting of miRNAs expression can be achieved by using antisense inhibitor oligonucleotides (AMO) technique. AMOs are artificially designed oligonucleotides fully complementary to their target miRNAs, which act to breakdown the targeted miRNAs with unknown mechanisms. AMOs are a necessary tool for studying miRNAs and have also been proposed as potential therapeutic agents56, 57. The function of a series of AMOs has been investigated in cardiovascular diseases, and demonstrated favorable therapeutic results28, 31, 36. The application of AMOs, to some extent, possesses similar characteristics to that of gene therapy.
Delivery method is a key obstacle that must be overcome before AMOs can be used clinically. To increase the tissue uptake of AMOs, cholesterol moieties that facilitate cell penetration can be attached to AMOs to form “antagomiRs”, which can strongly inhibit the expression of target miRNAs after in vivo application58. More strikingly, the studies have validated the therapeutic effects of antagomiR in cardiac hypertrophy28 and infarction40, 41, 50 by intravenous bolus injection, indicating its promising clinical application perspective.
As one miRNA may act on multiple targets, its inhibitor AMO can also influence the expression of a large number of proteins leading to non-gene-specific action and “off-target” effects. To tackle this problem, a technique termed miRNA-masking antisense oligodeoxynucleotides (miR-Mask) was developed55, which was validated by testing its application to the cardiac pacemaker channel encoding genes HCN2 and HCN4. One of the major advantages of this technology is that it offers a gene-specific miRNA-interfering strategy. A miR-Mask is designed to be exactly antisense to the binding site for a given miRNA in the target mRNA, which can form duplex with the target mRNA. In this way, the miR-Mask acts as a target protector masking the binding site to stop the action of that miRNA. A recent study59 confirmed the efficiency of this technology in which the authors investigated the role of zebrafish miR-430 in regulating expression of TGF-Nodal agonist squint and antagonist lefty, the key regulators of mesendoderm induction and left-right axis formation. They designed target protector morpholinos complementary to miRNA binding sites in target mRNAs in order to disrupt the interaction of specific miRNA-mRNA pairs. Protection of squint or lefty mRNAs from miR-430 resulted in enhanced or reduced Nodal signaling, respectively59.
Another innovative approach termed “miRNA sponges” was invented by Ebert et al60. The idea behind it is to produce a single piece of RNAs containing multiple, tandem-binding sites for a miRNA seed family of interest, in order to target all members of that miRNA seed family, taking advantage of the fact that the interaction between miRNA and target is nucleated by and largely dependent on base pairing in the seed region (positions 2–8 of the miRNA). The authors constructed sponges by inserting tandemly arrayed miRNA binding sites into the 3'UTR of a reporter gene encoding destabilized green fluorescent protein driven by the cytomegalovirus promoter, which can yield abundant expression of the competitive inhibitor transcripts. Binding sites for a particular miRNA seed family were perfectly complementary in the seed region with a bulge at positions 9–12 to prevent RNA-interference-type cleavage and degradation of the sponge RNA. When vectors encoding these miRNA sponges are transiently transfected into cultured cells, they depress miRNA targets as strongly as the conventional AMOs. The major advancement of this technique over the AMO technique is that it can better inhibit functional classes of miRNAs than do AMOs that are designed to block single miRNA sequences.
It has become clear that a particular condition may be associated with multiple miRNAs, eg miR-29 and miR-21 in cardiac fibrosis and a given gene may be regulated by multiple miRNAs. Thus, simultaneously targeting multiple miRNAs relevant to a particular condition may offer an improved approach than targeting a single miRNA. To investigate this possibility, we developed an innovative strategy, the multiple-target AMO (MT-AMO) technology, which confers a single AMO fragment the capability of targeting multiple miRNAs61. We have validated the technique with two separate MTg-AMOs: anti-miR-21/anti-miR-155/anti-miR-17-5p and antimiR-1/anti-miR-13361. The MTg-AMO targeting miR-21, miR-155 and miR-17-5p produced a greater inhibitory effect on cancer cell growth, compared with the regular single-target AMOs. Moreover, while using the regular single-target AMOs excluded HCN2 as a target gene for either miR-1 or miR-133, the MTg-AMO approach is able to reveal HCN2 as the target for both miR-1 and miR-13361.
Perspectives and limitations
miRNAs are a group of key regulators that modulate the development of various diseases, including cardiovascular diseases. Some individual miRNAs can dictate the pathogenesis of a specific disease, such as cardiac hypertrophy, arrhythmias, ischemia and atherosclerosis, etc. Moreover, manipulating the expression of the disease-causing miRNAs can prevent the disease progression, strongly indicating the therapeutic potential of miRNAs. Furthermore, several miRNA manipulation techniques have been created to accomplish “gain-of-function” or “loss-of-function” management on miRNA expression. The constructs designed based on these techniques have all demonstrated their efficacy in treating cardiovascular disease, which makes miRNAs step closer to clinical therapeutic application. Thus, it is not surprising that miRNAs have been continually attracting attention of researchers from nearly every areas of life sciences, which will inevitably impel the progress in miRNA biology and therapy. Additionally, miRNAs have also demonstrated their potential as diagnostic and prognostic markers62. Recent evidence supports the use of specific miRNA signatures to predict clinical outcomes of relevant conditions62. Our pervious work has validated miR-1 as a proarrhythmic miRNA in ischemic heart disease31. In a recent study, we found that the serum miR-1 level is closely linked to the severity of cardiac infarction and the occurrence of lethal arrhythmias in coronary artery disease patients (unpublished data), which represents a new diagnostic marker.
However, we have merely made a first step towards the application of miRNA-interfering technologies. We still face many unanswered questions and unsolved problems. First, as a particular disease condition may be associated with multiple miRNAs, and a given gene may be regulated by multiple miRNAs. Thus, interactions between miRNAs may have impact on the protein expression of the target genes and may complicate the potential role of miRNA as therapeutic targets in cardiovascular disease. We should further validate the specificity of the established miRNA targets before application. Second, the detailed molecular mechanism under which miRNAs regulate cardiovascular diseases still requires further eludicitation. Third, the biggest obstable on the way to miRNA-based clinical therapy is how to efficiently deliver miRNA mimics and inhibitors to the target organs. Not until we will have had rational answers to these questions after rigorous fundamental and clinical studies, will we have better ideas about miRNAs as targets for the development of therapeutic agents for cardiovascular disease.
References
Lai EC . Micro RNAs are complementary to 3'UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 2002; 30: 363–4.
Wightman B, Ha I, Ruvkun G . Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993; 75: 855–62.
Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000; 408: 86–9.
Chua JH, Armugam A, Jeyaseelan K . MicroRNAs: biogenesis, function and applications. Curr Opin Mol Ther 2009; 11: 189–99.
Li M, Marin-Muller C, Bharadwaj U, Chow KH, Yao Q, Chen C . MicroRNAs: control and loss of control in human physiology and disease. World J Surg 2009; 33: 667–84.
Wang Z, Luo X, Lu Y, Yang B . miRNAs at the heart of the matter. J Mol Med 2008; 86: 771–83.
Latronico MV, Catalucci D, Condorelli G . Emerging role of microRNAs in cardiovascular biology. Circ Res 2007; 101: 1225–36.
Latronico MV, Condorelli G . MicroRNAs and cardiac pathology. Nat Rev Cardiol 2009; 6: 419–29.
Lee Y, Jeon K, Lee JT, Kim S, Kim VN . MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 2002; 21: 4663–70.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23: 4051–60.
Kim VN . MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol 2004; 14: 156–9.
Peters L, Meister G . Argonaute proteins: mediators of RNA silencing. Mol Cell 2007; 26: 611–23.
Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R . MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 2005; 7: 719–23.
Brennecke J, Stark A, Russell RB, Cohen SM . Principles of microRNA-target recognition. PLoS Biol 2005; 3: e85.
Nilsen TW . Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet 2007; 23: 243–9.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB . Prediction of mammalian microRNA targets. Cell 2003; 115: 787–98.
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006; 126: 1203–17.
McKinsey TA, Olson EN . Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest 2005; 115: 538–46.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN . Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007; 316: 575–9.
van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 2006; 103: 18255–60.
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 2007; 116: 258–67.
Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 2007; 42: 1137–41.
Sucharov C, Bristow MR, Port JD . miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol 2008; 45: 185–92.
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M . MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 2007; 100: 416–24.
Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol 2009; 29: 2193–204.
da Costa Martins PA, Bourajjaj M, Gladka M, Kortland M, van Oort RJ, Pinto YM, et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008; 118: 1567–76.
Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, et al. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 2007; 170: 1831–40.
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med 2007; 13: 613–8.
Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 2009; 119: 2772–86.
Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF . miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA 2009; 106: 12103–8.
Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 2007; 13: 486–91.
Anderson ME, Mohler PJ . MicroRNA may have macro effect on sudden death. Nat Med 2007; 13: 410–1.
Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE, et al. MiR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2 . Circ Res 2009; 104: 514–21.
Shan H, Zhang Y, Lu Y, Pan Z, Cai B, Wang N, et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res 2009; 83: 465–72.
Goette A . Nicotine, atrial fibrosis, and atrial fibrillation: do microRNAs help to clear the smoke? Cardiovasc Res 2009; 83: 421–22.
Luo X, Lin H, Pan Z, Xiao J, Zhang Y, Lu Y, et al. Down-regulation of miR-1/miR-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem 2008; 283: 20045–52.
Xiao J, Luo X, Lin H, Zhang Y, Lu Y, Wang N, et al. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem 2007; 282: 12363–7.
Luo X, Xiao J, Lin H, Li B, Lu Y, Yang B, et al. Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol 2007; 212: 358–67.
Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007; 129: 303–17.
van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 2008; 105: 13027–32.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008; 456: 980–4.
Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res 2009; 82: 21–9.
Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 2009; 104: 170–8, 6p following 178.
Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci 2007; 120: 3045–52.
Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem Biophys Res Commun 2009; 381: 597–601.
Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX, Lin SG, et al. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem Biophys Res Commun 2008; 376: 548–52.
Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B, et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischemic cardioprotection. Cardiovasc Res 2009; 84: 434–41.
Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C . MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol 2009; 47: 5–14.
Yin C, Wang X, Kukreja RC . Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett 2008; 582: 4137–42.
Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009; 324: 1710–3.
Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 2009; 119: 2357–66.
Zhang C . MicroRNAs: role in cardiovascular biology and disease. Clin Sci (Lond) 2008; 114: 699–706.
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res 2007; 100: 1579–88.
Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C . A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res 2009; 104: 476–87.
Xiao J, Yang B, Lin H, Lu Y, Luo X, Wang Z . Novel approaches for gene-specific interference via manipulating actions of microRNAs: examination on the pacemaker channel genes HCN2 and HCN4. J Cell Physiol 2007; 212: 285–92.
Chan JA, Krichevsky AM, Kosik KS . MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65: 6029–33.
Cheng AM, Byrom MW, Shelton J, Ford LP . Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005; 33: 1290–7.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature 2005; 438: 685–9.
Choi WY, Giraldez AJ, Schier AF . Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science 2007; 318: 271–4.
Ebert MS, Neilson JR, Sharp PA . MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 2007; 4: 721–6.
Lu Y, Xiao J, Lin H, Bai Y, Luo X, Wang Z, et al. A single anti-microRNA antisense oligodeoxyribonucleotide (AMO) targeting multiple microRNAs offers an improved approach for microRNA interference. Nucleic Acids Res 2009; 37: e24.
Vandenboom Ii TG, Li Y, Philip PA, Sarkar FH . MicroRNA and cancer: tiny molecules with major implications. Curr Genomics 2008; 9: 97–109.
Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program; 2007CB512000/2007CB512006) awarded to Prof Bao-feng YANG.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pan, Zw., Lu, Yj. & Yang, Bf. MicroRNAs: a novel class of potential therapeutic targets for cardiovascular diseases. Acta Pharmacol Sin 31, 1–9 (2010). https://doi.org/10.1038/aps.2009.175
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.175
Keywords
This article is cited by
-
MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3β
Acta Pharmacologica Sinica (2021)
-
Characteristics of Volcanic Reservoirs and Hydrocarbon Accumulation of Carboniferous System in Junggar Basin, China
Journal of Earth Science (2021)
-
Characteristics of Diagenesis and Pore Evolution of Volcanic Reservoir: A Case Study of Junggar Basin, Northwest China
Journal of Earth Science (2021)
-
miR32-5p promoted vascular smooth muscle cell calcification by upregulating TNFα in the microenvironment
BMC Immunology (2020)
-
Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFβ1 and miR-29 pathways
Scientific Reports (2016)
