
Abstract
We report here that some patients affected by schizencephaly are heterozygous for mutations in EMX2, a homeobox gene implicated in the patterning of the developing forebrain. Schizencephaly is a very rare human congenital disorder characterized by a full-thickness cleft within the cerebral hemispheres. Large portions of these may be absent and replaced by cerebrospinal fluid. We previously reported the presence of EMX2 mutations in 7 out of 8 sporadic cases of schizencephaly. We now extend this analysis to 10 additional patients, including 2 brothers. Six patients were found to be heterozygous for de novo mutations in EMX2. In particular, the 2 brothers show the same mutation affecting the splicing of the first intron, while this mutation is absent in their parents and in the 2 unaffected siblings.
Similar content being viewed by others
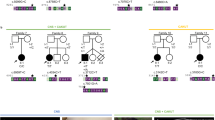


Schizencephaly [1, 2] is a rare congenital defect of the cerebral cortex, involving a full-thickness cleft of the cerebral hemispheres with consequent communication between the ventricle and pericerebral subarachnoid spaces. These clefts are characterized by the so-called pial-ependymal seam, an infolding of polymicrogyric gray matter along the cleft from the cerebral cortex into the ventricle [1–3]. In 80% of cases the septum pellucidum is absent and septo-optic dysplasia is observed in about one-third of cases [3]. The clinical signs of schizencephaly vary considerably, depending on the separation of the cleft lips and the position and size of the area involved [4]. Based on the space separating the walls of the fissure, an open-lip form and a closed-lip form can be distinguished. Though most of the clefts are bilateral in nature, those present in milder forms tend to be asymmetrical and often appear unilateral. Patients with mild forms of schizencephaly are often almost normal but may show partial epileptic seizures and mild spastic hemiparesis [2]. Conversely, patients with bilateral open-lip clefts usually have microcephaly, severe developmental delay with serious mental retardation and spastic quadriparesis [4].
We previously studied [5] 8 sporadic patients and their families and reported in 7 of them the occurrence of heterozygous mutations in the homeobox gene EMX2. This gene is known to control the development of the cerebral cortex and in particular the proliferation and possibly the migration of cortical neuroblasts [6–8]. We now performed single-strand conformation polymorphism (SSCP) analysis on PCR amplification products generated from genomic DNA of 10 additional patients (table 1), including 2 brothers (fig. 1), with appropriate oligonucleotides that span the entire coding region contained in the three exons of EMX2 [5]. These oligonucleotides, already used in previous experiments [5], are schematically shown in figure 2a. Oligonucleotides E2.5 and E2.6 detected altered fragments corresponding to second exon e2 in 6 of these patients, namely DZ, MB, MI, MM, PB and VF, whereas no alteration in the migration pattern of the various fragments has been found in 4 patients.

Magnetic resonance imaging (MRI) in patients with severe (a, b), intermediate (c) and mild (d) schizencephaly. a Patient MM: coronal T1-weighted image (w.i.) demonstrating a bilateral open-lip frontoparietal schizencephaly associated with corpus callosum hypoplasia. b Patient MI: axial T1-w.i. demonstrating a very large bilateral open-lip temporoparietal schizencephaly associated with frontal lobe agenesis and absence of the corpus callosum. c Patient DZ: coronal T2-w.i. demonstrating the open-lip left schizencephaly (arrowheads) associated with corpus callosum agenesis. A right closed-lip schizencephaly surrounded by heterotopic gray matter is also present in this patient (not shown). d Patient MB: coronal T2-w.i. demonstrating the unilateral right parietal closed-lip schizencephaly (arrowheads).

Mutations detected in EMX2. a Schematic representation of the EMX2 gene with the indication of the oligonucleotides used in SSCP analysis [5]. Boxes indicate the coding region subdivided in three exons, e1–e3. Stippled boxes indicate the two regions of the homeobox, separated by the second intron i2 at the level of codon 54. Introns are not represented to scale. b, c Sequence of the mutated region of EMX2 in patients MI, MM, VF and DZ (b) and in patients MB and PB (c). Substitutions and deletions are indicated in bold. Capical letters indicate the exonic sequence of second exon e2 and homeobox sequences are underlined. Arrowheads indicate the 3′ splice site of first intron il (b) and the 5– splice site of second intron i2 (c). d Nucleotide sequence of the relevant portion of the amplified DNA fragments from the 6 patients carrying a mutation.
Let us consider first patients MI and MM, 2 brothers. They showed severe, clearly bilateral schizencephaly, associated with agenesis/hypoplasia of corpus callosum. MI also shows partial frontal lobe agenesis. Sequencing of amplified DNA segments revealed in both the presence of the same mutation, namely aG→T substitution in position 1 of exon e2 (fig. 2b, (fig. 2d). This substitution changes the identity of the corresponding amino acid residue from glycine to valine if the splicing of the first intron occurs correctly, but it is also likely to interfere with the completion of this splicing. To test this hypothesis, we trans-fected both NIH3T3 cells and human embryonal carcinoma NT2/D1 cells with an expression vector containing the relevant region of either the mutated or wild-type form of EMX2 and analyzed its expression (fig. 3). RNase protection analysis (fig. 3b) failed to detect any properly spliced mRNA in cytoplasm of both cell types transfected with the mutated form. Neither the parents not two unaffected siblings of MI and MM carry this mutation.

Expression analysis of EMX2 genes present in patient MM in transfected NIH3T3 and NT2/D1 cells, a Schematic representation of the EMX2 region inserted in the pSG5 [12] expression plasmid. It includes the last 85 bp of the first exon el, the entire i1 intron, and the first 66 bp of the second exon e2. The probe used in RNase protection experiments is also shown. It represents 151 bp of the spliced mRNA. b RNase protection analysis [5] of cytoplasmic RNA from NIH3T3 cells transfected with the wild-type (wt) construct and with the mutated form (T1) and from NT2/D1 cells transfected with the mutated form (T2). No mature cytoplasmic RNA (fragment of 151 nucleotides) is present in cells transfected with the mutated form, while present in cells transfected with the wild-type construct. P = Probe; U = untransfected NIH3T3 cells.
Patient DZ showed severe, clearly bilateral schizencephaly, associated with agenesis of corpus callosum and of septum pellucidum. Analysis of this patient revealed the presence of an A→G substitution in position 15 of the homeobox (fig. 2b, d). This is a synonymous substitution leaving unaffected the identity of the corresponding amino acid residue, an arginine in position 5 of the homeodo-main. It was impossible to test the genotype of the parents, both unknown. We previously reported [5] the presence of a synonymous substitution for the arginine residue in position 3 of the homeodomain in three unrelated patients affected by mild schizencephaly. We found exactly the same mutation, a C→A substitution in position 7 of the homeobox, in patient VF, a case of mild, seemingly unilateral schizencephaly, associated with partial epilepsy and mild mental retardation (fig. 2b, d). The same mutation is present in the mother, as was the case for the previous patients with this mutation [5]. Finally, patients MG and PB carried point mutations in the second intron i2, consisting of a deletion and a substitution, respectively (fig. 2c, d). Both patients showed a mild, seemingly unilateral schizencephaly, associated with partial epilepsy. Both mutations were de novo mutations absent in more than 1,500 chromosomes of control individuals.
The patients analyzed can be subdivided into two distinct clinical groups (table 1) [2–9]. Four of them show a severe, clearly bilateral schizencephaly, associated with more or less conspicuous malformations of the forebrain and causing severe hemiparesis (Fa) or tetraparesis (MI, MM, DZ), while 6 of them show mild, seemingly unilateral schizencephaly, associated with partial epilepsy and causing mild hemiparesis with negligible or mild (VF) mental retardation. Partial epilepsies always mapped to the cortical region of the schizencephalic cleft. We detected mutations in EMX2 in 3 out of 4 severe cases (fig. 2b, d). Two of these patients (MI and MM) are brothers and show the same mutation, a substitution in position 1 of the second exon resulting in a splicing defect. The observed substitution is absent in their parents and in the 2 unaffected siblings. This suggests a case of gonadal mosaicism in either one of the parents. The 3rd patient of this group, DZ, carries a synonymous substitution in the fifth amino acid residue of the homeodomain. It is difficult to hypothesize any pathological role for this synonymous mutation, as was the case for the 3 examples of synonymous mutations in EMX2 previously reported [5]. Further analysis of the transcriptional or posttranscriptional properties of these mutated forms is clearly required, as some synonymous mutations have recently been reported [10] to result in altered gene products. In this line, it may be of interest to note that the nucleotide substitution found in patient DZ created a palindromic sequence (fig. 2b) absent in normal individuals.
We also detected mutations in EMX2 in 3 of the 6 analyzed patients of the second group (fig. 2b, d) (table 1), but the sequence alterations observed cannot be associated with a clear mechanism for either loss of function or gain of abnormal function. Whereas patients MB and PB showed point mutations in the second intron, patient VF carried a C→A synonymous substitution in the third amino acid residue of the homeodomain. This mutation occurs within the same palindromic sequence of that found in patient DZ and is identical to that already described [5] in 3 unrelated patients affected by a mild, unilateral schizencephaly, which was clinically indistinguishable from that observed in VF. Thus, this same mutation has been identified so far in 4 out of 13 EMX2 mutations observed in schizencephaly, while absent in more than 1,500 controls. These findings render more and more unlikely that this is a chance occurrence, even if it is conceivable to hypothesize that we are observing a second site mutation which might follow a first major mutation event. Experiments are underway to understand the pathogenesis in these patients. This analysis cannot be performed in the affected tissues and the very restricted expression pattern of this gene prevents simple assays in different tissues of the affected people. Therefore, we are currently producing transgenic mice carrying these mutations. It should be also reminded that severe molecular defects in this gene are probably incompatible with survival, due to the severity of the corresponding cerebral and urogenital defects, observed in corresponding animal models [8–11].
In conclusion, our data are compatible with a role of mutations in the gene EMX2 in at least some cases of schizencephaly and a case may now be made for a correlation between the molecular nature of these mutations and the observed clinical severity. In fact, predictably deleterious molecular defects, like frameshift mutations or mutations affecting the splicing pattern are invariably associated with severe, open-lip, bilateral schizencephaly, whereas subtle or leaky mutations are associated with mild, closed-lip, seemingly unilateral schizencephaly (table 1). From the practical point of view, our data disclose the possibility to devise a molecular differential diagnosis to distinguish schizencephaly from other similar cortical dysplasias and to further discriminate between different clinical forms of schizencephaly.
References
Wolpert SM, Barnes PD: MRI in Pediatric Neuroradiology. St. Louis, Mosby Year Book, 1992.
Granata T, Battaglia G, D’Incerti L, Franceschetti S, Spreafico R, Savoiardo M, Avanzini G: Schizencephaly: Clinical findings; in Guerrini R, Andermann F, Canapicchi R, Roger J, Zifkin BJ, Pfanner P (eds): Dysplasias of Cerebral Cortex and Epilepsy. Philadelphia, Lippincott-Raven, 1996.
Barkovich AJ, Norman D: MR imaging of schizencephaly. AJR 1988;150:1391–1396.
Barkovich AJ, Kjos B: Schizencephaly: Correlation of clinical findings with MR characteristics. AJNR 1992;13:85–94.
Brunelli S, Faiella A, Capra V, Nigro V, Simeone A, Cama A, Boncinelli E: Germline mutations in the homeobox gene EMX2 in patients with severe schizencephaly. Nature Genet 1996;12:94–96.
Simeone A, Gulisano M, Acampora D, Stornaiuolo A, Rambaldi M, Boncinelli E: Two vertebrate homeobox genes related to the Drosophila empty spiracles gene are expressed in the embryonic cerebral cortex. EMBO J 1992;11: 2541–2550.
Gulisano M, Broccoli V, Pardini C, Boncinelli E: Emx1 and Emx2 show different patterns of expression during proliferation and differentiation of the developing cerebral cortex. Eur J Neurosci 1996;8:1037–1050.
Pellegrini M, Mansouri A, Simeone A, Boncinelli E, Gruss P: Dentate gyrus formation requires Emx2. Development 1996;122:3893–3898.
Granata T, Battablia G, D’Incerti L, Franceschetti S, Spreafico R, Savoiardo M, Avanzini G: Schizencephaly: Neuroradiologic and epileptogenic findings. Epilepsia 1996;37:1185–1193.
Richard I, Beckmann JS: How neutral are synonymous codon mutations? Nature Genet 1995;10:259.
Miyamoto N, Yoshida M, Kuratani S, Matsuo I, Aizawa S: Defects of urogenital development in mice lacking Emx2. Development 1997; 124: 1653–1664.
Green S, Issemann I, Sheer E: A versatile in vivo and in vitro eukaryotic expression vector for protein engineering. Nucl Acids Res 1988; 16:369.
Acknowledgments
We are indebted to A. Cama, G.G. Consalez, R. Guerrini and L. Wrabetz for a number of helpful comments and suggestions. This work was supported by grants from the Mariani Foundation for Pediatric Neurology, EC BIOTECH and BIOMED Programmes, the Telethon-Italia Programme and the Italian Association for Cancer Research (AIRC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Faiella, A., Brunelli, S., Granata, T. et al. A Number of Schizencephaly Patients Including 2 Brothers Are Heterozygous for Germline Mutations in the Homeobox Gene EMX2. Eur J Hum Genet 5, 186–190 (1997). https://doi.org/10.1007/BF03405915
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF03405915
