
Abstract
In early 1992, the European bumblebee, Bombus terrestris, was first seen in Tasmania and currently has spread to most of the island. Here, we report on the genetic structure, using micro-satellites, of the invading population from samples collected in the years 1998–2000, a few years after the first sighting of the species in its new area. The data show that the Tasmanian population has a very low genetic diversity, with less than half of the allelic richness (Richness=2.89 alleles; Hexp=0.591) and lower levels of heterozygosity as compared to populations in New Zealand (4.24 alleles; Hexp=0.729) and Europe (5.08 alleles; Hexp=0.826). In addition, the genetic data suggest that the invasion must have happened once, probably around late 1991, and was the result of very few, perhaps only two, individuals arriving in Tasmania. Furthermore, these founders came from the New Zealand population. Today, the population in the south of Tasmania seems to act as a source population from which individuals migrate into other parts of the state. A similar source–sink structure seems also the case for New Zealand. The data show that B. terrestris is a highly invasive species capable of establishing itself even after a dramatic genetic bottleneck. B. terrestris may be an invasive species due to the haplo–diploid sex determination system, which exposes recessive, deleterious mutations to selection. Offspring of such purged lines may then be able to tolerate high levels of inbreeding.
Similar content being viewed by others

Introduction
Invasion by alien species into an existing biota is associated with many ecological problems of today. The process itself has many ramifications for the biology of resident species, ecological communities and not the least for the invader itself. For example, invasion by the zebra mussel (Dreissena polymorpha) into North America is associated with a reduction in plankton organisms (Pace et al., 1998) and the alteration of macro-invertebrate communities and competing mussel populations (Schloesser et al., 1996; Strayer et al., 1996). Different but no less severe problems have been caused by invasive social insects such as the Red Imported Fire Ant (Solenopsis invicta) spreading in North America (Porter and Savignano, 1990), the Little Fire Ant (Wasmania auropunctata) invading the Galapagos islands (Lubin, 1984), or the Africanized honey bee (Apis mellifera scutellata) that has steadily expanded its range from its origin in Brazil to the recent invasion of the southern US where it creates difficulties for honey bee breeders and crop pollination (Winston, 1992). Why an invasion is successful typically depends on a number of ecological and genetic factors (Nentwig, 2006). For example, the invader may have an increased tolerance against chemo-physical conditions, a broad diet, high reproductive capacity (Essink and Dekker, 2002), or may have left its competitors and enemies behind (Colautti et al., 2004; Prenter et al., 2004). In social insects, plasticity in the organization of social structures (Tsutsui and Suarez, 2003), or differences in the rate of colony development (Schneider et al., 2004) could provide additional benefits when adapting to a new environment.
However, invasions are presumably often based on few individuals, in which case the effects of genetic drift become particularly prevalent, potentially leading to a strongly homozygous population of closely related individuals. By definition, such populations attain high degrees of inbreeding; inbreeding in turn is typically associated with fitness losses (Keller and Walter, 2002). More recently, invasions became better studied also at the genetic level (Lee, 2002; Astanei et al., 2005; Colautti et al., 2005; Lindholm et al., 2005; Stepien and Tumeo, 2006), in particular to elucidate the most likely sources of the invasion and ways of spread. Here, we report on an initial study of the invasion genetics of a social insect in Tasmania.
In early 1992, specimens of the European bumblebee, Bombus terrestris (subfamily Bombini, Apidae, Hymenoptera), were first recorded in Battery Point near the dock area of Hobart, Tasmania (Semmens et al., 1993). Originally, the subfamiliy Bombini does not occur in the geographical region southeast of Indonesia. However, in the course of European settlement, several Bombus species were purposefully introduced into New Zealand. A first successful establishment of introduced bumblebees was recorded in 1885 and repeated in 1906 in the Canterbury plains around Christchurch. Since then, no more introductions of Bombus species have been attempted in New Zealand (Hopkins, 1914; Thompson, 1922; Macfarlane and Gurr, 1995). Bumblebees are highly valued pollinators worldwide and substantially add to the value of crop production (Goulson, 2003). Their introduction into New Zealand was therefore regarded a high priority in the 19th century, as the newly landed English farmers relied on meat production and thus on sufficiently high-quality cattle feed, which was also introduced with red clover (it was, in fact, Charles Darwin who pointed out the importance of bumblebees as prime pollinators of red clover). The attraction to introduce particularly useful bumblebee species, such as B. terrestris, into farm areas outside Europe has not weakened since then. The fact that B. terrestris suddenly appeared in Tasmania – isolated from the nearest source populations of New Zealand by many thousand miles of open ocean – therefore rekindled the speculations about a possible purposeful introduction. Alternatively, bumblebee queens accidentally could have travelled on freight ships travelling between New Zealand and Tasmania. Queens in fact are quite capable of shutting down their activities and remain quiescent in the cold and for many days even outside the usual hibernation cycle. They so could easily travel, for example, on board of ocean going vessels for considerable distances. Regardless, the introduced population in Tasmania has now spread over most of the island (Hingston et al., 2002), and the species seems to thrive without any too obvious effects of inbreeding depression (Hingston, 2005).
Here, we report on the results of genetic screening, based on micro-satellites, of the invading population of B. terrestris in Tasmania, their likely source population in New Zealand and a comparison with an extant population in England, from where the first introductions into this part of the world were made 100 years ago. Our aim was to test that the introduced population is indeed genetically impoverished, that it would originate from New Zealand and whether it has invaded with only a few individuals.
Materials and methods
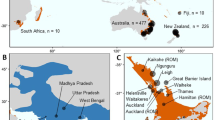
Specimens (diploid workers and queens) of B. terrestris were sampled in Tasmania in the seasons of 1999 (20–22 February) and 2000 (1 February to 6 April, but most before mid-March), and those from New Zealand in the season of 1998 (15–25 January; Figure 1). Sampling in Tasmania and New Zealand was carried out at points with bumblebee habitats separated several kilometres apart, which makes it very unlikely that individuals from the same colony are sampled (Chapman et al., 2003) (who report maximum foraging distances of 0.7–3.2 km for B. terrestris). Additional specimen from England was collected in June 2005. Sample points and number of specimen analysed per locality are given in Figure 1 and Table 1, respectively. For the core analysis, we genotyped a total of 63 individuals from New Zealand, 169 individuals from Tasmania (sampled in 2000) and 24 from Lincolnshire, England. In addition, we had typed 49 individuals sampled from two locations in the pilot sample from Tasmania in 1999, which are included here for full information.

Map of sampling locations in Tasmania (left) and New Zealand (right). The scale (horizontal bars) indicates 100 km in each case.
The genetic screening was performed at seven micro-satellite loci (eight loci in Tasmania 1999). The respective sequences, primers and technical protocols have been published elsewhere (Estoup et al., 1993; Allander and Schmid-Hempel, 2000). The screening resulted in a data set containing the alleles at each locus and for each sampling location. This data set was analysed with a number of different software packages as mentioned in more detail in Results. We treated New Zealand, Tasmania and England as regions with several populations (except England).
Results
Table 1 shows the allelic richness and levels of heterozygosity found in the samples, using FSTAT (Estoup et al., 1996). Overall, the degree of expected heterozygosity (Hexp, which corrects for sample size) was consistently smaller in Tasmania as compared to New Zealand (Table 1), but the values per population or locus did partly overlap. Similarly, the allelic richness (FSTAT, based on a minimum of four individuals) in Tasmania (sample of year 2000; average richness=2.89 alleles) was lower than in the New Zealand population (4.24 alleles; P=0.002, with 1000 permutations). For Tasmania in 1999, allelic richness was an average of 2.89 alleles (Hexp=0.55). We found a single instance of one allele (170 bp at Locus B11) that only occurred in Tasmania (a queen in 1999 at site ‘Hobart’, a worker each at ‘Hobart’ and ‘South’, 2000) and which was not found in New Zealand. All other Tasmanian alleles were contained in the New Zealand samples.
Using Genepop on the web (Raymond and Rousset, 1995), the observed allele frequencies were in Hardy–Weinberg for Tasmania 1999 (X2=12.173, df=4, P=0.59), Tasmania 2000 (X2=51.308, df=54, P=0.58) and in New Zealand (X2=88.499, df=72, P=0.091). In Tasmania also no deviation was found when each locus and each population were considered separately. In the New Zealand populations, significant deviation from Hardy–Weinberg was observed in two instances (at Locus B100 in population Christchurch with P=0.042; at Locus B126 in population Otago with P=0.021).
We used an analysis of molecular variance implemented in Arlequin ver. 3.01 (Excoffier et al., 2005) to partition the observed genetic variation. The analysis suggested that 86.7% of the total variation is contained within the populations (sum of squares (SS): 85.138, df=2, variance component (var)=0.2994); another 11.2% results from differences among regions (SS: 41.753, df=10, var=0.0521); while little effect was found for the variation among populations within region (1.95%; SS: 1159.134, df=499, var=2.3229) (number of diploids typed n=64, 164 and 24 for New Zealand, Tasmania and England, respectively). This pattern was confirmed with additional analyses. For example, using Genepop to calculate FST values, the Tasmanian and New Zealand populations were little differentiated from each other (Table 2). Population South from Tasmania in 1999 showed a lower value of FIS than expected by chance (FIS=−0.137, P=0.028).
In the following, we report only results using the data Tasmania 2000 because of its more detailed coverage as compared to the Tasmanian sample of 1999. When so compared, the overall genetic differentiation (New Zealand vs Tasmania 2000) was FST=0.139 with an FIS=−0.008. The genetic structure of B. terrestris was additionally investigated without a priori population information with a factorial correspondence analysis on alleles performed by GENETIX (ver. 4.01b) (Belkhir et al., 1996) (Figure 2). The program STRUCTURE is less reliable to identify clusters without any a priori assumptions (Pritchard et al., 2000). However, using its Bayesian clustering method (full admixture model used here) STRUCTURE also recovered the three regions as the most likely number of population clusters among the individual genotypes (graph not shown). Indeed, there are clear biological reasons to suspect that England, New Zealand and Tasmania indeed form three distinct populations. Based on these clusters, STRUCTURE can perform assignment tests by determining the probability of each individual genotype to occur in a given cluster. On the population level, only the sample from Ruatapu (New Zealand) could have belonged either to England or New Zealand.

Clustering of individual genotypes based on a factorial correspondence analysis using GENETIX. This analysis does not take into account a priori population information; it clustered New Zealand individuals apart from Tasmania, with the former overlapping somewhat with Lincolnshire.
To gain a deeper insight into the phylogeographic structure of the sample, the population of England was considered the ancestral stock to all bees in the Australia – New Zealand region. With this root, we reconstructed the phylogeny with micro-satellite allele frequencies using PHYLIP (Felsenstein, 1989). Frequencies were transformed into pairwise (Nei's) genetic distances between populations with the program ‘Gendist’ of the PHYLIP package. These were then converted into a phylogenetic tree with program ‘Neighbor’ (the neighbour-joining method). Finally, we bootstrapped the data with ‘Seqboot’ (using 1000 replicates) and constructed a consensus tree with ‘Consensus’ (Figure 3). This analysis again grouped the New Zealand populations among themselves and apart from Tasmania, as well as apart from England (Lincolnshire).

Consenus phylogenetic tree of the populations of Tasmania (2000) and New Zealand (1998). The tree is rooted with the Lincolnshire (England) population. Constructed with PHYLIP 3.61 using the NJ-UPGMA algorithm based on micro-satellite allele frequencies. The numbers at forks indicate the number of times the group comprise the species which are to the right of that fork occurred among the trees, out of 1000 replicated trees (using Seqboot). Populations in bold are from New Zealand (see Figure 1).
We further used the package BayesAss (Bayesian Assignment) (Wilson and Rannala, 2003) to estimate recent migration rates (that is, over the last few generations) between pairs of populations based on the genotypes of individuals; this procedure uses a Bayesian approach with Markov chains and Monte Carlo techniques (10 million iterations of which the first 5 million were used as a burn-in). Hence, in contrast to the phylogenetic tree (Figure 3), this analysis allows to estimate the current exchange of genes among populations in ecological time – in the case of Tasmania a short time after the first arrival of B. terrestris. We applied this procedure to the New Zealand and Tasmanian populations and found that both were fully separated from each other, that is, with no evidence of recent gene flow. Within each region, there was evidence for a source–sink system. In Tasmania, population ‘South’ acted as a source and the same was the case for ‘Otago’ in New Zealand. Depending on starting conditions, ‘Kaikoura’ sometimes acted as the New Zealand source in the calculations; this instability in the result is most likely due to the smaller sample sizes or lack of sufficient differentiation in New Zealand. It was consistent, however, that migration rates were less than 2% for all other pairs of locations, a value conventionally considered to be non-relevant. Also when we compared pooled Tasmania and pooled New Zealand populations, no recent migration was evident between the two regions (into New Zealand from Tasmania: m=0.010±0.009; vice versa: m=0.002±0.001).
Finally, we applied coalescence analysis to assess the size of the founding population in Tasmania. In particular, we used the isolation with migration model with changing population size (modified from Hey, 2005) implemented in the Bayesian-based IM program (Nielsen and Wakeley, 2001) to estimate the population parameters for B. terrestris. Assuming a stepwise mutation model for the evolution of variation at micro-satellite loci, IM estimates the likelihood function of the demographic parameters backward in time. In our case, this extends back to the time when Tasmanian population split from its presumed shared ancestral New Zealand population. Given the nonsignificant global FST values among New Zealand and Tasmanian populations and the result of the Structure analysis, all populations within New Zealand (n=63) and Tasmania (n=169), respectively, were pooled. With this procedure, the demographic parameter estimates are, for New Zealand, Tasmania and the total population, respectively, the effective population sizes in coalescence units (ΘNZ, ΘTS and ΘA), the gene flow rates (from New Zealand to Tasmania, mNZ; and vice versa, mTS) and the population splitting time (t). From these estimates, the corresponding demographic values (NNZ, NTS and NA) can be gained by scaling with the neutral mutation rate (μ) weighed by generation time. The parameter, s, is the fraction of the ancestral population (NA) that forms the New Zealand population (NNZ), that is the fraction 1−s gives rise to the Tasmanian population (NTS) (Table 3). The IM analyses were conducted using a burn-in time of 100 000 steps followed by 1 million simulations and a Metropolis coupling of two Markov chains with a two-step increment model as described in the IM documentation (Hey and Nielsen, 2006). The posterior probability distributions for the parameters of the first run were used to adjust the scalar of parameters for two additional runs with different random number seeds. To convert these results to demographic units we estimated mutation rate, μ, as μ=t/t with t being the (floating) coalescent time and by assuming a time, t, since splitting of 10 years (that is, from the likely first occurrence of B. terrestris in Tasmania until our samples were taken in 2000); the generation time is set to 1 year. This resulted in an average of μ=6 × 10−4 (range 10−5…11 × 10−4). This mutation rate fits well within the range of 6.5 × 10−5 to 6.6 × 10−3 observed in honeybees (Apis mellifera; Moritz et al., 2003).
Estimated coalescence parameters for the split between the two bumblebee populations are summarized in Table 3. The coalescent estimate for the shared ancestral population is θA=6.39, implying an effective population size of very few individuals connecting New Zealand and Tasmanian populations (translating into an ancestral total population size of NA≈2700 individuals; Table 3). The estimate of the fraction of the ancestral population founding today's New Zealand population (s=0.999) in turn suggests the Tasmanian population goes back to very few animals from the ancestral population – with an estimated (1−s)NA≈3 individuals. The estimate of the present effective population size for Tasmania is NTS=29 (Table 3). These estimates are consistent with a strong expansion of the bumblebee population since its establishment in Tasmania.
Discussion
The successful invasion of Tasmania by B. terrestris (Hingston et al., 2002; Hergstrom et al., 2005) could have been greatly facilitated by the loss of parasites in the process (Allen et al., 2007), as well as by the generalized diet of the species that allowed utilizing introduced and native flowering plants alike (Hingston and McQuillan, 1998; Hingston et al., 2002; Hingston, 2006). Yet a remarkable characteristic of the Tasmanian population is the impoverished allelic richness as compared to New Zealand or Europe (Table 1) (Estoup et al., 1996; Widmer et al., 1998). Genetic impoverishment is also suggested by a high frequency (ca. 50%) of laboratory-bred colonies producing diploid males in a sample of queens raised a few years after the introduction (Buttermore et al., 1998). This high frequency indicates an invading population even lower than our estimate of not more than three queens. Therefore, the population must be quite robust against the possible effects of low genetic diversity and associated inbreeding regardless of its potential ecological success otherwise. A reason, ironically, could be the haplo–diploid sex determination system of hymenoptera, which (in haploid males) leads to a strong effect of purging selection against deleterious alleles. This should leave some families with above-average genotypes; in fact, at least in the context of immune defences, tolerance to inbreeding was observed in laboratory tests with B. terrestris (Gerloff and Schmid-Hempel, 2005). It may be those high-quality families whose offspring are able to invade a new area even with a small founder population. Furthermore, the invading population in Tasmania seems to have left its parasites behind (Allen et al., 2007), among other things, it lacks the trypanosome Crithidia bombi, which drastically lowers the success of founding queens in spring (Brown et al., 2003) and which is known to occur in New Zealand (Macfarlane et al., 1995).
The low diversity also suggests a severe bottleneck for B. terrestris during and after its introduction to the island. This is supported by the estimates from the coalescent analysis indicating a founding population of ca. three individuals and a substantial expansion (by a factor of 10) afterwards (Table 3). The genetic data are moreover compatible with the New Zealand population being ancestral to Tasmania. This follows from all our analyses showing the structure and mutual relationships of populations (Figures 2 and 3) and the fact that virtually all alleles of Tasmania are also represented in New Zealand. Given the bootstrap values (Figure 3), the split between Tasmania and New Zealand-Lincolnshire is real. The East population in Tasmania (ranging over a wide area, from Dodges Ferry to Orford; roughly 40 km as the crow flies, or 70 km by land) appears somehow separated from the rest of this region. These findings fit the recorded history of the species with an early introduction from Europe to New Zealand 100 years ago and a subsequent introduction into Tasmania. The earlier introduction from England occurred in two waves and was based on larger population sizes (Hopkins, 1914; Thompson, 1922; Macfarlane and Gurr, 1995). Accordingly, the genetic diversity of the New Zealand populations is comparable to those found in native European populations (Table 1).
Furthermore, gene flow between New Zealand and Tasmania either over recent generations (from BayesAss) or since the split (coalescent, IM; 2 Nm values ≪1, Table 3) is not present – hence, the introduction was in all likelihood a single event. The first sightings of B. terrestris in Tasmania were on 19 February 1992 and our (primarily analysed) samples are from 2000, thus, there are approximately 10 generations between the two dates (perhaps, B. terrestris might be partially bivoltine in these areas, that is a part of the population may have two generation cycles within a year). Had there been migration events since then, they should have left traces in our ‘BayesAss’ analyses. We thus propose that B. terrestris was introduced in a single episode, probably in 1991 or early in 1992, and that this must have involved very few, probably only two, fertilized queens. The actual invasion could have happened by accidental transport on board of commercial vessels leaving New Zealand for a port in Tasmania in late 1991. Enquiries with Hobart Ports Corporation (G Denney, personal communication) indicate that there were seven direct shipping arrivals at Hobart from New Zealand in the six months preceding the first sighting of B. terrestris in Tasmania. Private arrivals of yachts would not be recorded though. On the other hand, there were direct flights operating 1–2 times per week between Christchurch, New Zealand, and Hobart in the period immediately preceding the first discovery of the bumblebee in Tasmania (T Malmgren, Qantas Historical Collection, Sydney, Australia). Prior to the recorded presence of B. terrestris in 1992, there had been two quarantine interceptions in Hobart of B. terrestris on New Zealand–Hobart flights; one in the passenger cabin in 1983 and one from an unspecified location on the aeroplane in 1988 (Department of Primary Industry and Water, DPIW, Tasmania collection records, C Young, personal communication). An invasion via regular passenger flights therefore is a possibility. However, the historical records of the actual sightings (see Figure 4) show that the place of the first appearance was Battery Point, near the dock area of Hobart on the western shore of the Derwent River. Probably then, the mouth of the Derwent River had acted as an initial barrier to its eastern spread such that the spread from Hobart was towards the South and West during the early years (Figure 4) (Semmens et al., 1993; Hergstrom et al., 2005). If the bees arrived via Hobart airport at all, which is on the eastern side of the Derwent River some 20 km from Battery Point, they most likely were displaced towards Battery Point and released there.

Expansion of B. terrestris in Tasmania plotted from historical records until the summer of 2000–2001. Sources are DPIW records, University of Tasmania collections (Semmens et al., 1993; Buttermore, 1997; Hingston et al., 2002; Hergstrom et al., 2005). Symbols of sightings indicate the records of observations in new areas at the time. Lines show the approximate front of the invasion over the years. Dashed oval lines circle the areas where B. terrestris is abundant and firmly established. First sighting was in early 1992 in Hobart.
Populations, both, in Tasmania and New Zealand were following Hardy–Weinberg frequencies and showed little genetic differentiation among sampling locations. According to the estimate for recent migration events, this observation is probably due to a source-sink dynamics that should homogenize gene frequencies over populations within Tasmania and New Zealand in the years since the invasion. Similarly, large panmictic populations have been observed for mainland Europe (Estoup et al., 1996; Widmer et al., 1998). In Tasmania, population South (corresponding to the area south and southwest of Hobart) seems to send out migrants that dominate all other sampled populations. In New Zealand, this role pertains to Otago (or perhaps Kaikoura). In both cases, these areas are settled with many gardens and agricultural lands. It is therefore likely that these (genetic) source populations reflect productive habitats where B. terrestris is able to thrive on flowering plants that it can utilize efficiently.
Regardless of the many details that must remain speculative, our genetic analysis shows that B. terrestris in Tasmania is most likely of New Zealand origin and invaded with a very small founder population. Its success in colonizing Tasmania also demonstrates that B. terrestris can be a highly invasive species well able to cope with inbreeding but that this probably requires the fortunate above-average families to have their descendants arriving first.
References
Allander K, Schmid-Hempel P (2000). Immune defense reaction in bumble bee workers after a previous challenge and parasitic co-infection. Funct Ecol 14: 711–717.
Allen GR, Seeman OD, Schmid-Hempel P, Buttermore RE (2007). Low parasite loads accompany the invading population of the bumblebee, Bombus terrestris in Tasmania. Insectes Sociaux 54: 56–63.
Astanei I, Gosling E, Wilson JM, Powell E (2005). Genetic variability and phylogeography of the invasive zebra mussel, Dreissena polymorpha (Pallas). Mol Ecol 14: 1655–1666.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996). GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS: UMR 5171, Université de Montpellier, France.
Brown MJF, Schmid-Hempel R, Schmid-Hempel P (2003). Strong context-dependent virulence in a host–parasite system: reconciling genetic evidence with theory. J Anim Ecol 72: 994–1002.
Buttermore RE (1997). Observations of successful Bombus terrestris (L. Hymenoptera: Apidae) colonies in Southern Tasmania. Aust J Entomol 36: 251–254.
Buttermore RE, Pomeroy N, Hobson W, Semmens T, Hart R (1998). Assessment of the genetic base of Tasmanian bumble bees (Bombus terrestris) for development as pollination agents. J Apicult Res 37: 23–25.
Chapman RE, Wang J, Bourke AFG (2003). Genetic analysis of spatial foraging patterns and resource sharing in bumble bee pollinators. Mol Ecol 12: 2801–2808.
Colautti R, Ricciardi A, Grigorovich I-A, Macisaac H-J (2004). Is invasion success explained by the enemy release hypothesis? Ecol Lett 7: 721–733.
Colautti RI, Manca M, Viljanen M, Ketelaars HEM, Bürggi A, Macisaac HJ et al. (2005). Invasion genetics of the Eurasian spiny waterflea: evidence for bottlenecks and gene flow using microsatellites. Mol Ecol 14: 1869–1879.
Essink K, Dekker R (2002). General patterns in invasion ecology tested in the Dutch Wadden Sea: the case of a brackish-marine polychaetous worm. Biol Invasions 4: 359–368.
Estoup A, Solignac M, Cornuet JM, Goudet J, Scholl A (1996). Genetic differentiation of continental and island populations of Bombus terrestris (Hymenoptera: Apidae) in Europe. Mol Ecol 5: 19–31.
Estoup A, Solignac M, Harry M, Cornuet J-M (1993). Characterization of (GT)n and (CT)n microsatellites in two insect species: Apis mellifera and Bombus terrestris. Nucleic Acids Res 21: 1427–1431.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetic analysis. Evol Bioinformatics Online 1: 47–50.
Felsenstein J (1989). PHYLIP – phylogeny interference package (Version 3.6). Cladistics 5: 164–166.
Gerloff CU, Schmid-Hempel P (2005). Inbreeding depression and family variation in a social insect, Bombus terrestris (L*) (Hymenoptera, Apidae). Oikos 111: 67–80.
Goulson D (2003). Conserving wild bees for crop pollination. Food Agric Environ 1: 142–144.
Hergstrom K, Buttermore R, Hopkins A, Brown V (2005). The distribution and spread of the bumblebee Bombus terrestris (L.) in Tasmania since introduction in 1991, with notes on foodplants. Kanunnah 1: 103–125.
Hey J (2005). On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol 3: 0965–0975.
Hey J, Nielsen R (2006). IM documentation. http://lifesci.rutgers.edu/~heylab/HeylabSoftware.htm#IM.
Hingston AB (2005). Inbreeding in the introduced bumblebee Bombus terrestris causes uncertainty in predictions of impacts on native ecosystems. Ecol Manag Restor 6: 151.
Hingston AB (2006). Is the exotic bumblebee Bombus terrestris really invading Tasmanian native vegetation? J Ins Cons 10: 289–293.
Hingston AB, Marsden-Smedley J, Driscoll DA, Corbett S, Fenton J, Anderson R et al. (2002). Extent of invasion of Tasmanian native vegetation by the exotic bumblebee Bombus terrestris (Apoidea: Apidae). Aust Ecol 27: 162–172.
Hingston AB, McQuillan PB (1998). Does the recently introduced bumblebee Bombus terrestris (Apidae) threaten Australian ecosystems? Aust J Ecol 23: 539–549.
Hopkins I (1914). The history of the introduction of bumble bees to New Zealand. NZ Dept Agric 2: 28pp.
Keller LF, Walter DM (2002). Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241.
Lee CE (2002). Evolutionary genetics of invasive species. Trends Ecol Evol 17: 386–391.
Lindholm AK, Breden F, Alexander HJ, Chan W-K, Thakurta SG, Brooks R (2005). Invasion success and genetic diversity of introduced populations of guppies Poecilia reticulata in Australia. Mol Ecol 14: 3671–3682.
Lubin YD (1984). Changes in the native fauna of the Galapagos Islands following invasion by the little red fire ant, Wasmania auropunctata. Biol J Linn Soc 21: 229–242.
Macfarlane RP, Gurr L (1995). Distribution of bumble bees in New Zealand. NZ Entomol 18: 29–36.
Macfarlane RP, Lipa JJ, Liu HJ (1995). Bumble bee pathogens and internal enemies. Bee World 76: 130–148.
Moritz RFA, Scharpenberg H, Lattorff HMG, Neumann P (2003). A technical note for using microsatellite DNA analysis in haploid male DNA pools of social hymenoptera. Insectes Sociaux 50: 398–400.
Nentwig W (ed). (2006). Biological Invasions. Springer: Berlin.
Nielsen R, Wakeley J (2001). Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158: 885–896.
Pace ML, Findlay SEG, Fischer D (1998). Effects of an invasive bivalve in the zooplankton community of the Hudson River. Freshwater Biol 39: 103–116.
Porter SD, Savignano DA (1990). Invasion of polygyne fire ants decimates native ants and disrupts arthropod community. Ecology 71: 2095–2106.
Prenter J, MacNeil C, Dick JTA, Dunn AM (2004). Roles of parasites in animal invasions. Trends Ecol Evol 19: 385–390.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Raymond M, Rousset F (1995). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Schloesser D, Nalepa T, Mackie GL (1996). Zebra mussel infestation of unionid bivalves (Unionidae) in North America. Am Zool 36: 300–310.
Schneider S-S, Degrandi-Hoffman G, Roan Smith D (2004). The African honey bee: factors contributing to a successful biological invasion. Annu Rev Entomol 49: 351–376.
Semmens TD, Turner E, Buttermore R (1993). Bombus terrestris (L) (Hymenoptera, Apidae) now established in Tasmania. J Aust Entomol Soc 32: 346.
Stepien CA, Tumeo MA (2006). Invasion genetics of Ponto–Caspian gobies in the Great Lakes: a ‘cryptic’ species, absence of founder effects, and comparative risk analysis. Biol Invasions 8: 61–78.
Strayer DL, Powell J, Ambrose P, Smith LC, Pace LM, Fischer DT (1996). Arrrival, spread and early dynamics of a zebra mussel (Dreissena polymorpha) population in the Hudson River estuary. Can J Fish Aquat Sci 53: 1143–1149.
Thompson GM (1922). The Naturalisation of Animals and Plants in New Zealand. Cambridge University Press: Cambridge.
Tsutsui ND, Suarez AV (2003). The colony structure and population biology of invasive ants. Conserv Biol 17: 48–58.
Widmer A, Schmid-Hempel P, Estoup A, Scholl A (1998). Population genetic structure and colonization history of Bombus terrestris s.l. (Hymenoptera: Apidae) from the Canary Islands and Madeira. Heredity 81: 563–572.
Wilson GA, Rannala B (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Winston ML (1992). Killer Bees – The Africanized Honey Bee in the Americas. Harvard University Press: Cambridge, MA.
Acknowledgements
Petra Ramseier helped with the laboratory work and genetic typing; Ben Sadd provided bees from England. We thank Jody Hey for his help with the IM program, and Ross Crozier for hosting PSH and RSH during 1998–1999. Financially supported by a Grant from the Swiss NSF (no. 3100-066733 to PSH), ETH (no. 0-20-010-95 to PSH), ARC small grant (no. A0011199 to GRA) and a University of Tasmania sabbatical grant to GRA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schmid-Hempel, P., Schmid-Hempel, R., Brunner, P. et al. Invasion success of the bumblebee, Bombus terrestris, despite a drastic genetic bottleneck. Heredity 99, 414–422 (2007). https://doi.org/10.1038/sj.hdy.6801017
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801017
Keywords
This article is cited by
-
Multiple sources implicated in the red swamp crayfish invasion in Michigan, USA
Biological Invasions (2023)
-
Invasion genetics of the Asian hornet Vespa velutina nigrithorax in Southern Europe
Biological Invasions (2022)
-
Evidence for multiple introductions of an invasive wild bee species currently under rapid range expansion in Europe
BMC Ecology and Evolution (2021)
-
Genetics, morphology and diet of introduced populations of the ant-eating Texas Horned Lizard (Phrynosoma cornutum)
Scientific Reports (2019)
-
Lack of genetic structuring, low effective population sizes and major bottlenecks characterise common and German wasps in New Zealand
Biological Invasions (2019)
