
Abstract
We examined intraspecies genetic variation in house mice (Mus musculus molossinus) from the northern third of the Japanese Islands, in order to obtain evidence of the history of mouse colonization that might have shaped the current genetic diversity. We extended the previous sampling of mitochondrial cytochrome b sequence and added information from the Y-linked Sry gene and ribosomal RNA gene surveys. We distinguish mitochondrial haplotypes characteristic of the North Asian musculus subspecies group (involving M. m. musculus and M. m. molossinus) as ‘MUS’, and that of the Southeast Asian castaneus subspecies group as ‘CAS’ (although the mice resemble MUS morphologically). There was a clear geographic partition of MUS and CAS types into southern and northern Hokkaido, respectively. Conversely, on Tohoku, the MUS and CAS types were interspersed without clear geographic subdivision. In contrast to the mtDNA data, all Hokkaido and Tohoku mice examined were found to possess a unique type for the Y-linked Sry gene, specific to Korea and Japan. Restriction site analysis of nuclear rDNA probe showed a consistent distribution of MUS and CAS types, as major and minor components, respectively, in the Hokkaido and Tohoku mice. These data support the previous notion that the Hokkaido and Tohoku mice experienced genetic hybridization between primary residents of CAS origin and MUS newcomers arriving via a southern route. The invasion of the MUS type could correspond with the evidence for arrival of prehistoric peoples. There are, however, alternative interpretations, including genetic admixture between MUS arriving by a southern route and CAS from a northern route.
Similar content being viewed by others


Introduction
The way in which genetic diversity is generated and maintained is highly variable among species and perhaps even subtly unique for each species (Avise, 2000). The house mouse (Mus musculus) is a particularly fascinating subject for demographic and evolutionary studies, due to its extensive and polytypic natural distribution, its long-term association with people (Boursot et al, 1993) and propensity for accidental, long-distance dispersal. In addition, the biomedical importance of M. musculus provides the impetus for ongoing studies of its ecology, morphology, cytology and genetics (Boursot et al, 1993; Moriwaki et al, 1994; Yonekawa et al, 2003). However, despite these efforts, our knowledge of this species remains inadequate in many respects. For example, there are still many unanswered questions regarding the time and place of origin of the various phylogenetic lineages of house mice (variously treated in recent literature as subspecies, full species or ‘semispecies’), the timing and routes of dispersal of these lineages, and the subsequent mode of interaction among the lineages, where they meet in secondary contact. Even more fundamentally, we are still seeking the most appropriate combinations of markers by which to characterize the genetic structure and determine the evolutionary history of this uniquely fascinating species.
M. musculus is today one of the most widely distributed of all mammals, with populations on all continents except Antarctica. Numerous, locally distinctive forms have been named, but recent taxonomic treatments generally group these into as many as 15, or as few as four subspecies, based on morphological traits of the skin and skull (Schwarz and Schwarz, 1943; Marshall, 1981, 1986). In contrast, molecular phylogenetic evidence favours a rather simpler view that divides M. musculus into three broadly distributed genetic groups and one or two additional groups with smaller geographic ranges (Britton and Thaler, 1978; Sage, 1981; Yonekawa et al, 1981, 1988; Ferris et al, 1983; Bonhomme et al, 1984; Suzuki et al, 1986; Auffray et al, 1991; Boursot et al, 1993; Moriwaki et al, 1994; Prager et al, 1998). We will refer to these principal groups as: DOM (M. m. domesticus) from Western Europe and North Africa; MUS (M. m. musculus) from the northern part of Eurasia except Western Europe; CAS (M. m. castaneus) from Southeast Asia; BAC (M. m. bactrianus) from India; and GEN (M. m. gentilulus) known so far only from Yemen.
The place of origin of the ancestral M. musculus stock is still hotly debated. Based on allozyme data sets, several research teams have proposed that M. musculus emerged in central Asia or India, and dispersed in both west and east, giving rise to DOM and CAS in Europe and East Asia, respectively (Boursot et al, 1996; Din et al, 1996). Under this scenario, the derived MUS and BAC types developed in situ, each in different areas of the Indian subcontinent. However, MUS types later dispersed out from this region, again moving both west and east, where they eventually established secondary contact with the DOM and CAS populations, respectively (Boursot et al, 1996; Din et al, 1996). An alternative view, derived from analyses of mitochondrial and some nuclear gene sequences, postulates a Middle Eastern origin of M. musculus, with subsequent dispersal of DOM, MUS, CAS and GEN types to Western Europe, northern Eurasia, India and the Arabian Peninsula, respectively (note that sequence data generally fail to identify a separate BAC cluster).
The zone of secondary contact between MUS and DOM lineages in Western Europe has attracted considerable interest, with detailed studies of chromosomes, allozymes, gene sequences, field ecology and reproductive behaviour (eg Boursot et al, 1993). In contrast, the distribution of MUS and CAS lineages in East Asia is only partially documented and there have been few studies of the consequences of secondary contact (Moriwaki et al, 1994). One notable exception is provided by the so-called ‘native’ Japanese population of M. musculus. On morphological criteria this population was long treated as a separate subspecies, M. m. molossinus. However, molecular phylogenetic studies using mitochondrial markers found no distinct evolutionary components in these populations at all, but rather a mix of elements derived from two ‘subspecies’ lineages – MUS from Northern China and CAS from Southeast Asia (Yonekawa et al, 1988, 2003). Initially, the geographic distribution of these components posed something of an enigma because the CAS type, usually associated with southern Asia, was restricted in Japan to Northeast Honshu (Tohoku district) and Hokkaido, both in the northern part of the island chain (Yonekawa et al, 1988), while the MUS type, otherwise found in northern Eurasia, was dominant in southern regions of Japan. To account for this unusual spatial distribution, Yonekawa et al (1988, 2003) proposed a hypothesis involving two phases of dispersal, followed by hybridization. Under this scenario, the Japanese islands were first colonized by mice of CAS type, perhaps via a southern route, with establishment of CAS populations on all islands. Sometime later, mice of MUS type arrived, probably via the Korean peninsula, leading to complete replacement of the CAS mitochondrial lineages in the southern and central islands, and an apparent admixture of CAS and MUS lineages in the northern parts. Although the mitochondrial data clearly point to a complex history of interaction between CAS and MUS lineages in Japan, maternal markers alone cannot answer many of the more interesting questions about how this interaction occurred. For example, did the CAS and MUS lineages hybridize with introgression of MUS haplotypes through most populations, or was there a true replacement of CAS by MUS populations? And if hybridization and introgression did occur, what can we learn from molecular sources about the pattern and process of interaction between the populations of different origin?
To shed more light on these issues, we characterized the genetic constitution of Hokkaido mice using a number of maternally, paternally and biparentally inherited genetic markers that might reveal in more detail the extent and sources of the genetic diversity. First, we examined the mitochondrial cytochrome b gene (cyt b: 1140 bp) to allow our specimens to be related to the previously defined subspecies lineages. The cyt b data also provide a means of estimating the time of divergence of the main mitochondrial lineages. We then analysed sequences of the Sry gene, which is located on the Y chromosome. Intraspecies variation in Sry has been used previously for subspecies-group recognition in M. musculus (Gubbay et al, 1992; Tucker et al, 1992; Nagamine et al, 1994; Lundrigan and Tucker, 1994; Albrecht and Eicher, 1997). As cyt b and Sry are not affected by meiotic recombination, these genes should allow us to trace the history of maternal and paternal lineages, respectively, and to identify any bias in the involvement of males and females in ‘hybrid’ coupling. In addition to these uniparentally inherited gene markers, we also examined restriction site variation in the biparentally inherited nuclear ribosomal RNA gene (rDNA) spacer (Suzuki et al, 1986, 1997; Iwasa and Suzuki, 2003). The rDNA gene is tandemly repeated, with several hundred copies per cell, and evolves under high homogenization pressure. Accordingly, blotting patterns generated for this marker are likely to provide reliable phylogenetic evidence, being resistant to degradation of phylogenetic signal, even after genetic hybridization. Suzuki et al (1990) found clusters of more than 10 rDNA loci in a previous study of rDNA in Asian mice; the observed variation also coincided well with independently assessed M. musculus subspecies groups (Suzuki et al, 1986). Our results support the notion that mice in Hokkaido and north Honshu have experienced genetic hybridization between MUS and CAS types; however, different spatial patterns are observed for each of the maternal, paternal and biparental markers, suggesting a complex history of interaction with different outcomes in each area.
Materials and methods
Materials
Mice were obtained from a variety of sources. Those from the principal study areas on Hokkaido and northeast Honshu were trapped in Sherman traps set around houses and in rice fields. Trap success was low, generally less than one mouse per 50 trap nights, and it was rare to catch more than one mouse in any specific locality. As the sample was accumulated over an extended period and from many different trap stations, it is unlikely that many of the captured individuals are immediate close relatives.
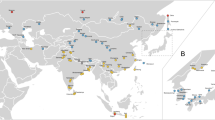
We studied DNA from a total of 66 mice from 36 localities (Table 1, Figure 1). In all, 26 mice came from Hokkaido, 17 from Northeast Honshu (Tohoku district) and 14 from other parts of Japan. The mice from Tohoku had previously been analysed for their mitochondrial D-loop and partial cyt b (264 bp) sequences (Furusawa, 2000) and we produced the complete cyt b sequence from five of these individuals. We also studied the DNA of seven mice from five localities outside Japan: Indonesia, Korea, Nepal, Russia and Taiwan. The following laboratory strains were obtained from the National Institute of Genetics, Mishima, Japan: SWN/Ms and CAS-Bog, originated from Sweon (Korea) and Bogor (Indonesia) respectively. Total DNA was extracted from tissues by the conventional phenol–chloroform method.

Collection localities and mitochondrial DNA types of the house mouse M. musculus from East Asia (left), with special emphasis on northern Japan (right). Locality numbers are identified in Table 1. Genotypes based on cyt b gene sequence variation are as follows: M. m. musculus (closed circle), M. m. castaneus (triangle for CAS I group, open circle for CAS II group), M. m. domesticus (closed square), and unknown type, ‘Nepal’, possibly M. m. bactrianus (open square). Genotypes based on restriction length polymorphism of the nuclear rDNAs are also shown with alphabetical letters; M. m. castaneus (C), M. m. musculus (M), and a mixed type of M. m. musculus and M. m. castaneus (M/c) as major and minor components, respectively.
Cyt b sequence analysis
The polymerase chain reaction and direct sequencing of the cyt b gene were performed according to previously described methods (Suzuki et al, 2004). The double-stranded PCR product was sequenced utilizing the PRISM Ready Reaction DyeDeoxy Terminator Cycle Sequencing Kit (ABI) and an ABI310 automated sequencer. The nucleotide sequences reported in this paper appear in the DDBJ, EMBL and GenBank nucleotide sequence databases under the accession numbers AB205273–AB205322.
Cyt b gene sequences were obtained from databases for M. m. castaneus (originated from Bangladesh), M. m. molossinus (strain MOA, originated from Aichi Prefecture, central Honshu, Japan), M. spicilegus (Suzuki et al, 2004) and M. m. domesticus (strains NZB and MilP; Loveland et al, 1990).
Sry sequence analysis
We amplified and determined sequences of the Sry gene in the downstream region of the 5′ end of the MHG box, using primer sets of Sry-8653 and Sry-9475 (Albrecht and Eicher, 1997; positions numbered according to the mouse reference sequence X67204; Gubbay et al, 1992). The reactions were carried out for 35 cycles, each consisting of 30 s at 94°C for denaturation, 30 s at 59°C for annealing and 90 s at 72°C for extension. These reaction mixtures (20 μl) contained 1.5 mM MgCl2. Sequencing of the PCR products was performed as described above.
Sry gene sequences were obtained from databases for two inbred strains: B6JEI-YPOS (Y derived from M. m. domesticus), CAST/Ei (Y derived from M. m. castaneus) and B6JEI-YCZECHII (Y derived from M. m. musculus) (Albrecht and Eicher, 1997). The sequences for M. spicilegus and M. spretus (Albrecht and Eicher, 1997) were used as outgroups.
Southern blot analysis for nuclear rDNA spacers
We used a probe of 0.7-kb mouse rDNA fragment that contained the 3′ end of the gene for 28S rRNA (Suzuki et al, 1986; Iwasa and Suzuki, 2003). The probe was prepared from an EcoRI-6.6-kb rDNA fragment, gel purified and digoxigenin-labelled (DIG labelling and detection kit, Boehringer Mannherim). Strand genomic DNA were digested with EcoRI restriction enzyme and electrophoresed through 0.7% agarose gels at 3 cm/V for 10–16 h in TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8.0). Capillary transfer of digested DNA to a nylon filter (Hybond-N; Amersham) was performed by the standard method. Immunodetection and visualization with the colorimetric substrates NBT/X-phosphate were performed according to the manufacturer's instructions.
Not all individuals were included in the Southern blot analysis. For some individuals, either we lacked DNA of sufficient quantity or quality to perform this analysis, or else we wished to conserve the samples for future, alternative uses. Our sampling strategy focused on establishing the geographic pattern for rDNA/RFLP in northern Japan, with less emphasis on other regions.
Phylogenetic data analysis
We constructed a phylogenetic tree by the neighbour-joining (NJ) method (Saitou and Nei, 1987) using genetic distances (d) computed by Kimura's two-parameter (K2P) method (Kimura, 1980) and performed bootstrap analysis with 1000 replicates. The robustness of the resulting phylogeny was also assessed by bootstrap analysis (100 replicates) using a maximum parsimony method. All computations were performed using PAUP 4.0b10 (Swofford, 2001).
Results
Cyt b sequences
We analysed complete cyt b haplotypes for 51 individuals of M. musculus (Table 1) and constructed a NJ tree for the 1140-bp sequences with M. spicilegus as outgroup (Figure 2). Three distinct clades are seen, representing the three subspecies groups of M. m. castaneus (CAS), M. m. domesticus (DOM), and M. m. musculus (MUS). One specimen from Nepal possessed a distinct haplotype, possibly representing the enigmatic M. m. bactrianus (Yonekawa et al, 1981; Moriwaki et al, 1994). A further 15 individuals represented by 264 bp of cyt b (Furusawa 2000) were allocated to clades during a subsequent analysis using the shorter fragment (Table 1). The geographic distribution of all haplotypes across the 36 localities is shown in Figure 1.

A NJ tree based on cyt b gene sequences. The tree was constructed with K2P model in consideration of all codon positions. Bootstrap scores obtained from NJ and maximum parsimony methods are expressed as percentages for each node. Only values greater than 50% are indicated and a dash ‘—’ means the value is lower than 50%. The major clades indicated in this tree are considered to have originated from the three subspecies groups of M. m. castaneus (CAS I and CAS II), M. m. domesticus (DOM), and M. m. musculus (MUS). The sequence of an individual from Nepal is discrete from the other types; it is here designated as ‘Nepal’ and may represent that of M. m. bactrianus.
Five distinct haplotypes are represented in the 26 mice from Hokkaido (Table 1). These haplotypes include examples of both the MUS group (two haplotypes, nine mice) and the CAS group (three haplotypes, 17 mice). The distribution of the two haplotype groups on Hokkaido is mutually exclusive; CAS group haplotypes occur throughout the northern part of the island (localities 7–14; Figure 1), while MUS group haplotypes occur in the southern region (localities 15–19). No locality produced both MUS and CAS group haplotypes. Among the 17 individuals with haplotypes of the CAS group from Hokkaido, 15 individuals shared the same haplotype; the remaining two each had unique haplotypes, which differed from the common haplotype by one and three bases (Otaru, HS2340 and Kushiro, KT3284), respectively. The geographic areas that support the CAS and MUS mtDNA populations are separated to some extent by the presence of an interposed mountainous region.
The 17 mice from Tohoku district in northern Honshu also possessed a mixture of MUS and CAS haplotype groups, with 13 from the MUS group and four from the CAS group (Table 1). The geographic distribution of the two groups (Figure 1) is interesting; as on Hokkaido, no locality produced both groups, but the spatial pattern is more complex. CAS group haplotypes seem to be confined to the eastern side of the island, where the MUS group is also present but uncommon (only recorded at Towa). Only MUS group haplotypes were found in the western and central regions of Honshu. The latter situation is consistent with other regions of Japan, where all studied populations possess MUS group haplotypes (Yonekawa et al, 1988).
All four Japanese CAS group haplotypes (CAS 3–6) belong to one major clade, here designated as CAS II subgroup (Figure 1). This clade also contains the CAS-Bog strain (CAS-2) originating from Bogor, West Java (differing from the common Japanese type, CAS-3, by two substitutions). A second major clade, designated CAS I subgroup, contains a single haplotype (CAS-1) from a Taiwanese mouse. CAS I and CAS II subgroups are quite divergent, with a total of 17 substitutions (maximum genetic distance d=0.016). The association of the Japanese and Indonesian haplotypes in CAS II, rather than with the Taiwanese haplotype of CAS I, is consistent with a notion that Japanese CAS group mice originated from somewhere in Southeast Asia rather than Taiwan or southern China; a comparable result was obtained from analysis of the D-loop sequences by Yonekawa et al (2003). An interesting detail is that the Tohoku CAS group haplotype (CAS-6) represented by complete cyt b sequence differ by one to two substitutions from the common CAS group haplotype (CAS-3) on Hokkaido.
The cyt b haplotypes from the MUS clade were less variable (genetic distance, d=maximum 0.005) and no geographic structure was found. The Korean mice from Peklyong Island and Sweon are almost identical in cyt b sequence to the MOA strain (originally from central Honshu) and to the majority of our sequences from Honshu and Hokkaido (MUS 2–7). Interestingly, a MUS group haplotype (MUS-8) was obtained from one individual from Okinawa Island. On the phylogenetic tree this haplotype is shown as the sister lineage to all other MUS haplotypes, but with a short common branch uniting all other members of this clade.
A surprising result was obtained for newly collected samples from Bogor, Java, that were identified on external and cranial morphology as M. m. castaneus. These were found to have typical European DOM group haplotypes (DOM 2 and 3) (Table 1, Figure 2). This finding contrasts with the CAS affinity of the laboratory strain that originated from Bogor (CAS-Bog) and raises the possibility of some recent admixture of DOM and CAS types in Java.
Sry sequences
The mouse Sry gene is known to show intraspecific variation in the downstream region from the 3′ end of the HMG box (Figure 3). Boissinot and Boursot (1997) discovered only two major haplotypes for Sry in the house mouse: one in DOM type mice and the other in both MUS and CAS type mice. The CAS+MUS Sry haplotype is distinguished from the DOM haplotype by substitutions at positions 8701, 8731 and 9019 (Lundrigan and Tucker, 1994; Albrecht and Eicher, 1997; positions as in Gubbay et al, 1992). In addition, substitutions occurring at position 8711 are specific to ‘molossinus’ type mice, with the C or T substitution at position 8711 associated with the presence or absence of TaqI site (Gubbay et al, 1992; Tucker et al, 1992; Lundrigan and Tucker, 1994; Nagamine et al, 1994). Nagamine et al (1994) revealed through their extensive geographic survey that the latter is confined to ‘molossinus’ mice from Japan and Korea.

A diagrammatic representation of the predicted Sry proteins from a M. m. musculus (Gubbay et al, 1992). Variant sites that are used for typing are shown with representative individuals. The outgroup sequence is the consensus of M. spicilegus and M. spretus.
For this study, we determined the Sry gene sequence (ca. 800 bp, Figure 3) for 33 individuals of M. musculus (Table 1). The Sry gene fragment showed highly variable sites with numerous triplet repeats (eg (CAG)n) in some regions. The Sry gene region spanning positions 8709–8777 was more stable, but showed informative substitutions at positions 8711 (C/T), 8721 (A/G), 8731 (C/T) and 8775 (C/T) (Figure 3). The Sry sequences from our study, combined with those previously reported, belong to a total of five haplotype groups. These are geographically restricted, as follows: A (Western Europe), B (Nepal), C (Indonesia), D (Taiwan/Russia) and E (Korea/Japan) (Figure 3). Haplotype E corresponds with the ‘molossinus’ type reported by previous authors, with nucleotide T at position 8711.
The consensus Sry gene sequence of the two outgroup species (M. spicilegus and M. spretus) is identical to haplotype D (Figure 3). It thus seems likely that haplotype D is the ancestral haplotype for M. musculus, which has been retained in each of the CAS and MUS lineages. Haplotype E differs from this by one substitution and could have arisen in either lineage.
In contrast to the strong geographic patterning found in the cyt b haplotypes, we found that the Sry gene sequences for the mice from Japan, including both Hokkaido and Tohoku, are monotypic: all belong to group E. Outside of Japan this haplotype is found only in Korea (Table 1). A male mouse from Bogor had a unique Sry haplotype that differs from that of a DOM-derived strain (B6JEI-YPOS) in one substitution (position 8775). This individual also had a DOM group haplotype for cyt b. The same substitution is also present in the Sry sequence of an individual from Nepal (Kathmandu, HS1523); however, this sequence also includes one additional substitution, at position 8721 (Figure 3). Russian (Kraskino, HS1473) and Taiwanese (Taitong, HS2400) individuals display Sry haplotype D, previously reported from M. m. castaneus and M. m. musculus, and represented in our data set by inbred strains CAST/Ei and B6JEI-YCZECHII respectively (Figure 3).
rDNA-RFLP
Each rDNA repeat unit is composed of a coding region and a nontranscribed spacer (NTS). The NTS is known to have a high rate of nucleotide substitutions that makes it suitable for examining intraspecific variation (eg Suzuki et al, 1986, 1997). We examined restriction-site variation of NTS in 31 individuals from Hokkaido, Tohoku district, Honshu, and other localities, using EcoRI restriction enzyme and the mouse 28S rDNA probe. Suzuki et al (1986) demonstrated that the EcoRI polymorphic sites downstream of the 5′ end of the 28S gene are specific to each population, as follows: DOM (6.0 kb only), MUS (9.0 kb only) and CAS/BAC (variable combination of 9.0, 7.5, 7, 6.5 kb, etc). The restriction patterns in mice from Hokkaido and Honshu are reported to share a 7.5-kb band, but with an additional 8.8-kb band as a minor component only in Honshu (Suzuki et al, 1986).
Eight individuals of M. musculus from four localities in Hokkaido showed the MUS type restriction patterns. Four of these individuals came from the northern area and thus possess mitochondrial CAS haplotypes, while four came from southern Hokkaido where MUS mitochondrial haplotypes occur (Table 1). In all, 12 individuals from eight localities on Hokkaido showed what appeared to be mixed MUS/CAS-type restriction patterns, with a minor 7.5-kb as well as the major 9-kb band. These localities are also distributed both north and south of the boundary between MUS and CAS mitochondrial types (Figure 1).
Populations of M. musculus on Honshu display a similar complex admixture of MUS and MUS/CAS restriction patterns (Table 1). Individuals with a minor 7.5-kb band were found throughout the northern part of Honshu, including localities where the mitochondrial cyt b haplotypes are exclusively of the MUS group (eg Sakata, Sakekawa, Tsuruoka). However, localities further south in central Honshu appear to lack the CAS contribution to the restriction pattern. Outside Japan, the 7.5-kb CAS band was detected in a mouse from Taiwan (Taitong), while a MUS pattern (9.0 kb only) was found in mice from Kraskino in Russia and Peklyong Island off the west coast of Korea. A faint band of 8.8 kb was exclusive to mice from Japan and Korea. This latter finding confirms the previous report of Suzuki et al (1986) and is also congruent with the geographic patterning of Sry haplotypes, wherein the Japanese and Korean samples possess the unique synapomorphic substitution from C to T at position 8711.
Discussion
Our results confirm previous reports that CAS mtDNA elements, diagnostic of Southeast Asian mice, are present in the northern part of Japan, Hokkaido and Tohoku (Yonekawa et al, 1988, 2003; Moriwaki et al, 1994). However, while previous studies found only CAS types in these areas, our larger data set clearly shows that MUS mtDNA elements are also present in both regions. Interestingly, in the case of Hokkaido mice, we found geographic separation between the two groups, with CAS group haplotypes restricted to northern localities and MUS types in the southern localities. In contrast, the Tohoku district of northern Honshu shows a somewhat more complex pattern: MUS group haplotypes are present throughout the region, while CAS types are restricted to the eastern part of Tohoku. Although no single locality produced mice with both haplotype groups, sample sizes in the eastern area are small and true mixed populations may yet be found.
The paternally inherited Sry and biparentally inherited rDNA-RFLP data sets each show different patterns to the mtDNA. The Sry gene is monotypic for the molossinus haplotype (type E in Figure 3) throughout Hokkaido and Tohoku, and also in more southern parts of Japan. In contrast, rDNA-RFLP indicates an admixture of pure MUS types and mixed MUS/CAS types in both regions (Figure 1). The latter result contrasts with the situation further south on Honshu, where a previous survey of rDNA-RFLP revealed MUS types exclusively (Suzuki et al, 1986). Our data thus support the notion that mice from Hokkaido and Tohoku are of mixed CAS/MUS origin. However, they indicate either a more complex genetic history involving distinct, area-specific demographic factors or a quite different immigration history to the current scenario.
The dual origin of house mice in northern Japan
The presence of CAS mtDNA in the northern third of Japan has been interpreted as a relict feature within the context of the hybrid-origin hypothesis (Yonekawa et al, 1988, 2003; Moriwaki et al, 1994). Under this hypothesis, the first mice to colonize Japan came from Southeast Asia, carrying CAS mtDNA types; these CAS mice spread throughout the Japanese islands. Subsequently, a second wave of immigration occurred, this time consisting of mice with MUS mtDNA types, presumably coming from China. The MUS mtDNA spread throughout the southern and central regions of Japan, completely replacing the original CAS mtDNA in most areas, but leaving a remnant pocket of CAS mtDNA in Hokkaido.
Our mtDNA data are broadly congruent with this historical scenario, although we have identified some new distributional features, namely the presence of MUS mtDNA in southern Hokkaido, and a pocket of CAS mtDNA in northeastern Honshu (Tohoku district). In addition, a dual-origin historical scenario is clearly supported by the rDNA-RFLP data that show the presence of the 7.5-kb band of rDNA, a marker for the CAS population, in mice from northern Honshu and Hokkaido. This marker is not present in mice from further south in Japan (Suzuki et al, 1986). The ubiquitous occurrence of the E haplotype of the Sry gene in Japan is less obviously consistent with a dual-origin hypothesis.
A dual-origin hypothesis for Japanese M. musculus suggests a possible parallel history with that of human populations in East Asia, including Japan (Tajima et al, 2002; Tanaka et al, 2004). As demonstrated by Tajima et al (2002), a total of six Y-chromosome haplotype groups are represented among Asian peoples. Three groups are dominant, representing north Asian (ht7 of Tajima et al, 2002), Han Chinese/southeast Asian (ht4) and exclusively Japanese (ht1) populations, respectively. Estimates of coalescence times for the haplotype groups range from 53 000 to 95 000 years, during the early period of dispersal of modern humans (ie Homo sapiens) (Tajima et al, 2002). Japanese people display all three of the common Y chromosome types, but with variable geographic representation. Among indigenous Ainu populations from Hokkaido, ht1 haplotypes are frequent and ht7 haplotypes are present as a minor component. In contrast, populations from Honshu and Kyushu have a high frequency of ht4 haplotypes, with ht1 and ht7 as lesser components. Further south, indigenous populations from Okinawa show all three haplotype groups, but with ht1 most frequent.
The genetic evidence for a dual origin of Japanese people is also consistent with archaeological scenarios for the colonization of Japan. These scenarios postulate an early immigration by Neolithic peoples of the Jomon culture, possibly around 12 000 years ago (Tanaka et al, 2004), followed by a more recent wave of immigration about 2300 years ago (Kitagawa, 2000). The Ainu and Okinawan indigenous populations are thought to be descendents of the earlier Jomon peoples, with populations on Honshu and Kyushu created by an admixture of the earlier and later peoples.
The combined genetic and archaeological evidence for two episodes of human migration to Japan, and most likely from different source areas, provides an obvious context for reconstruction of the migration history of M. musculus. However, any such reconstruction must deal with one intriguing observation – the fact that no remains of Mus have been found in a fossil or subfossil context in Japan (Kawamura, 1989). This issue is taken up again below.
Possible source areas for CAS and MUS founder populations
Yonekawa et al (2003) suggested from their study of mouse D-loop sequences that Japanese mtDNA haplotypes are more closely related to haplotypes from Southeast Asia rather than from southern China, including Taiwan. Our cyt b results appear to support this hypothesis: the CAS-Bog laboratory strain is very similar to the Japanese haplotypes, and these together are quite distinct from our Taiwanese sample. However, it must be cautioned that the Indonesian population itself probably represents a quite recent introduction from some other region.
Two lines of evidence point to a special relationship between the MUS populations of Japan and Korea. The first comes from the rDNA-RFLP dataset: a minor band of 8.8 kb is only recovered from MUS types in Korea and Honshu, implying a specific phylogenetic link between these populations. In addition, Japanese and Korean mice share the same derived haplotype of the Sry gene (type E). However, this haplotype is also present in the authentic inbred strains (eg BALB/c, C57B6), in which the Y chromosome is identified only as being of ‘Asian’ origin (Nagamine et al, 1994), thereby raising the possibility that Sry type E might be found in other areas.
More intensive geographic sampling is obviously needed before the source area of either component is identified with any confidence. A study currently underway using nuclear gene markers is expected to help resolve this issue.
Who arrived first – CAS or MUS?
As mentioned above, the currently favoured hypothesis for the origin of Japanese M. musculus suggests an initial colonization by CAS type mice. MUS types are seen as a later arrival, leading to widespread replacement of the original CAS types, except in the refugial area of northern Japan. However, two alternative scenarios need to be considered: (1) that MUS types arrived first and spread through the entire island chain, followed by a later invasion of CAS types into northern Japan, with local replacement of MUS types; and (2) MUS types and CAS types arrived around the same time, from the south and north, respectively, with establishment of several contact zones in Hokkaido and northeast Honshu. Each of these scenarios sheds a very different light on the genetic history of M. musculus in Japan.
The ‘CAS first’ scenario is in good agreement with the reconstructed history of human populations in Japan. However, as noted above, there is currently no evidence to suggest that M. musculus of any kind arrived with the earliest human migrants. Under this scenario, we need to pay special attention to two aspects of the genetic composition of Japanese mice. The first is the relatively sharp geographic separation of the mtDNA haplotypes in both Hokkaido and northeastern Honshu. Essentially, this region of northern Japan contains two ‘enclaves’ of CAS mtDNA, within a more general distribution of MUS types. The second is the absence of any indication of CAS contributions in the rDNA-RFLP profiles from anywhere in Japan outside the Hokkaido and Tohoku districts of Honshu. In particular, this observation needs to be reconciled with the evidence for extensive mixing of CAS and MUS rDNA-RFLP elements throughout northern Hokkaido, including areas where only CAS mtDNA is found. The presence of Sry haplotype E throughout Japan also needs to be accommodated within this scenario.
One possibility is that the observed genetic structure is the result of genetic drift, leading to a geographic subdivision of the mtDNA types of CAS and MUS populations in different areas. On Hokkaido, the northern (CAS) and southern (MUS) populations are partially isolated by a substantial mountainous region. However, in northeastern Tohoku district where the CAS mtDNA type is present, there is no obvious barrier between the CAS and MUS populations. If subspecies admixture between CAS and MUS populations occurred independently in each of Hokkaido and Tohoku through random drift, we might expect the rRNA gene also to show some differences between the two areas. However, this is not the case: the rDNA-RFLP profiles of Hokkaido and the Tohoku mice are extremely similar, including the presence of the faint 7.5-kb band. The rDNAs are known to be present on around 10 different chromosomes in Asian mice (Suzuki et al, 1990); hence, it is unlikely that such similar patterns would arise under a model of random drift. A more likely explanation of this similarity is that the Hokkaido and Tohoku populations are historically linked, perhaps by a dispersal event within the last few hundred years.
The presence of the derived Sry haplotype E throughout Japan also begs explanation. As noted earlier, this mutation could have arisen within a population of either MUS or CAS mice, since both of these lineages share the ancestral D haplotype. As independent origin of the E haplotype in both lineages is extremely unlikely, the ubiquity of this haplotype in Japan implies widespread and effective introgression from one lineage into the other.
The ‘MUS first’ scenario postulates an initial introduction of M. musculus with mtDNA of MUS type. Presumably, this population would also have possessed a rDNA-RFLP profile consisting of the single 9.0 kb band. Mice with such a genetic profile are indeed widespread in Kyushu, and in southern and central Honshu. Subsequently, under this scenario, mice of CAS type arrived in northern Japan. If they entered a region already occupied by MUS type genes, the currently observed genetic composition of Hokkaido and Tohoku mice would require a subsequent replacement in this area of MUS by CAS mtDNA types, but with an admixture of rDNA. This might imply either a strong selective mating of males of MUS type with females of CAS type, or a strong bias in the fitness of offspring with CAS mtDNA. However, irrespective of such mating or fitness biases, more effective introgression of biparentally inherited markers is to be expected. Not surprisingly then, the rDNA of CAS and MUS lineages appears to be thoroughly mixed throughout the northern populations. This contrasts markedly with the absence of rDNA of CAS type in populations of M. musculus in central and southern Honshu, where mtDNA types are exclusively MUS type. Effective introgression of the Sry E haplotype is also required under this scenario, but with the same uncertainty as to which lineage donated the derived Sry E haplotype.
The third scenario postulates that two groups of M. musculus colonized Japan around the same time, but coming from different source areas and arriving at different locations – MUS mtDNA types entering in the south, and CAS types in the north. Under this scenario, the contact zones between the two mtDNA types in each of northeast Honshu and Hokkaido might represent primary contact zones. Three features of the genetic composition of northern Japanese M. musculus would then require explanation, specifically the subsequent admixture of rDNA types throughout the northern part of Japan, the maintenance of discrete mtDNA populations and the broader introgression of the E haplotype of the Sry gene.
As noted above, effective introgression of rDNA types across a contact zone is expected given the ease of transfer of biparental markers. Provided some effective crossing occurs, rDNA components will rapidly become mixed along the immediate hybrid zone, accompanied by gene flow and introgression from there into each of the primary populations. This is precisely the situation observed in northern Japan, where CAS and MUS rDNA types are mixed over a broad area, extending throughout Hokkaido and penetrating south into central Honshu. Maintenance of the contact zone between CAS and MUS mtDNA is also readily explained through the inherent stability of such zones in the absence of any strong selective pressure for one of the two haplotype groups. This is due to the constant rarity of the invasive haplotypes in areas dominated by resident haplotypes, probably because of female philopatry and the presence of some geographic barriers; in Hokkaido, there is a chain of mountainous areas that demarcates the northern and southern groups of mtDNA. Differential dispersal by gender can further affect the relative magnitude of phylogeographical structure in biparentally and uniparentally inherited markers (Hare, 2001; Wen et al, 2004).
Complete introgression of Sry haplotype E throughout the range of M. musculus in Japan is more difficult to explain. One point to make at the outset is that we cannot be certain which of the postulated founder populations did in fact possess this derived Sry haplotype. It is even possible that transfer of the derived haplotype had occurred prior to entry of either lineage, in a source area where both MUS and CAS lineages were present, although this would need to have occurred without interchange of rDNA. However, assuming for the present that Sry haplotype E was present in one only of CAS- or MUS-type mice, we would need to find a mechanism to explain strong selection for transmission of one lineage of Y chromosomes over the other type. One possibility is that the replacement occurred as a consequence of random drift, as postulated earlier for the replacement of CAS by MUS mtDNA haplotypes under the ‘CAS first’ scenario. However, other possibilities are worthy of mention, including more effective long-distance dispersal of male mice across hybrid zones (Hare, 2001; Wen et al, 2004) and the possibility of differential production of abnormal sperm types in inter-subspecies hybrids involving males of CAS vs MUS Y chromosome type (Oka et al, 2004).
In terms of broader comparisons, it is of interest to note that the three different markers, maternal cyt b, paternal Sry and biparental nuclear rDNA, showed similar gene-specific patterns of spatial variation in Japanese dormice (Suzuki et al, 1997) and voles (Iwasa and Suzuki, 2003). It might be helpful to use even more, different gene markers to characterize population genetic structure and to assess the dynamics of hybrid interaction between closely related lineages. In particular, we may need many more markers for studies on organisms that have complicated evolutionary histories involving commensalism.
References
Albrecht KH, Eicher EM (1997). DNA sequence analysis of Sry alleles (subgenus Mus) implicates misregulation as the cause of C57BL/6J-YPOS sex reversal and difines the Sry functional unit. Genetics 147: 1267–1277.
Auffray J-C, Marshall JT, Thaler L, Bonhomme F (1991). Focus on the nomenclature of European species of Mus. Mouse Genome 88: 7–8.
Avise JC (2000). Phylogeography: The History and Formation of Species. Harvard University Press: Cambridge.
Boissinot S, Boursot P (1997). Discordant phylogeographic patterns between the Y chromosome and mitochondrial DNA in the house mouse – selection on the Y chromosome? Genetics 146: 1019–1034.
Bonhomme F, Catalan J, Britton-Davidian J, Chapman VM, Moriwaki K, Nevo E et al (1984). Biochemical diversity and evolution in the genus Mus. Biochem Genet 22: 275–303.
Boursot P, Auffray JC, Britton-Davidian J, Bonhomme F (1993). The evolution of house mice. Ann Rev Ecol System 24: 119–152.
Boursot P, Din W, Anand R, Darviche D, Dod B, Von Deimling F et al (1996). Origin and radiation of the house mouse: mitochondrial DNA phylogeny. J Evol Biol 9: 391–415.
Britton J, Thaler L (1978). Evidence for the presence of two sympatric species of mice (genus Mus) in southern France based on biochemical genetics. Biochem Genet 16: 213–225.
Din W, Anand R, Boursot P, Darviche D, Dod B, Jouvin-Marche E et al (1996). Origin and radiation of the house mouse: clues from nuclear genes. J Evol Biol 9: 519–539.
Ferris SD, Sage RD, Huang CM, Nielsen JT, Ritte U, Wilson AC (1983). Mitochondrial DNA evolution in mice. Genetics 105: 681–721.
Furusawa S (2000). Population Structure of the House Mouse from Northern Honshu, Japan Inferred from Maternally and Paternally Inherited Gene Marker. Master's Thesis, Yamagata University (in Japanese).
Gubbay J, Vivian N, Economou A, Jackson D, Goodfellow P, Lovell-Badge R (1992). Inverted repeat structure of the Sry locus in mice. Proc Natl Acad Sci USA 89: 7953–7957.
Hare MP (2001). Prospects for nuclear gene phylogeography. Trends Ecol Evol 16: 700–706.
Iwasa MA, Suzuki H (2003). Intra- and interspecific genetic complexities of two Eothenomys species in Honshu, Japan. Zool Sci 20: 1305–1313.
Kawamura Y (1989). Quaternary rodent faunas in the Japanese islands (Part 2). Mem Fac Sci Kyoto Univ Ser Geol Mineral LIV: 1–235.
Kimura M (1980). A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
Kitagawa Y (2000). Nonmetric morphological characters of deciduous teeth in Japan: Dachronic evidence of the past 4000 years. Int J Osteoarc 10: 242–253.
Loveland B, Wang CR, Yonekawa H, Hermel E, Lindahl KF (1990). Maternally transmitted histocompatibility antigen of mice: a hydrophobic peptide of a mitochondrially encoded protein. Cell 60: 971–980.
Lundrigan BL, Tucker PK (1994). Tracing paternal ancestry in mice, using the Y-linked, sex-determining locus, Sry. Mol Biol Evol 11: 483–492.
Marshall JT (1981). Taxonomy. In: Foster HL, Small JD, Fox JG (eds) The Mouse in Biomedical Research. Academic Press: New York. Vol 1, pp 17–26.
Marshall JT (1986). Systematics of the genus Mus. In: Potter M, Nadeau JH, Cancro MP (eds) The Wild Mouse in Immunology. Springer-Verlag: Berlin. pp 12–18.
Moriwaki K, Shiroishi T, Yonekawa H (1994). Genetics in Wild Mice. It's application to Biomedical Research. Japan Scientific Societies Press: Tokyo.
Nagamine CM, Shiroishi T, Miyashita N, Tsuchiya K, Ikeda H, Namikawa T et al (1994). Distribution of the molossinus allele of Sry, the testis-determining gene, in wild mouse. Mol Biol Evol 11: 864–874.
Oka A, Mita A, Sakurai-Yamatani N, Yamamoto H, Takagi N, Takano-Shimizu T et al (2004). Hybrid breakdown caused by substitution of the X chromosome between two mouse subspecies. Genetics 166: 913–924.
Prager EM, Orrego C, Sage RD (1998). Genetic variation and phylogeography of Central Asian and other house mice, including a major new mitochondrial lineage in Yemen. Genetics 150: 835–861.
Sage RD (1981). Wild mice. In: Forester HL, Small JD, Fox JG (eds) The Mouse in Biomedical Research. Academic Press: New York. Vol 1, pp 40–90.
Saitou N, Nei M (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Schwarz E, Schwarz H (1943). The wild and commensal stocks of the house mouse, Mus musculus Linnaeus. J Mammal 24: 59–72.
Suzuki H, Kurihara Y, Kanehisa T, Moriwaki K (1990). Variation in the distribution of silver-staining nucleolar organizer regions on the chromosomes of the wild mouse, Mus musculus. Mol Biol Evol 7: 271–282.
Suzuki H, Minato S, Tsuchiya K, Fokin IM (1997). Phylogenetic position and geographic differentiation of the Japanese dormouse, Glirulus japonicus, revealed by variations in rDNA, mtDNA and the Sry gene. Zool Sci 14: 167–173.
Suzuki H, Miyashita N, Moriwaki K, Kominami R, Muramatsu M, Kanehisa T et al (1986). Evolutionary implication of heterogeneity of the nontranscribed spacer region of ribosomal DNA repeating units in various subspecies of Mus musculus. Mol Biol Evol 3: 126–137.
Suzuki H, Shimada T, Terashima M, Tsuchiya K, Aplin K (2004). Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Mol Phylogenet Evol 33: 624–646.
Swofford DL (2001). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4. Sinauer Associates: Sunderland, MA.
Tajima A, Pan I-H, Fucharoen G, Fucharoen S, Matsuo M, Tokunaga K et al (2002). Three major lineages of Asian Y chromosomes: implications for the peopling of East and Southeast Asia. Hum Genet 110: 80–88.
Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N et al (2004). Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res 14: 1832–1850.
Tucker PK, Lee BK, Lundrigan BL, Eicher EM (1992). Geographic origin of the Y chromosome in ‘old’ inbred strains of mice. Mammal Genome 3: 254–261.
Wen B, Li H, Lu D, Song X, Zhang F, He Y et al (2004). Genetic evidence supports demic diffusion of Han culture. Nature 431: 302–305.
Yonekawa H, Moriwaki K, Gotoh O, Hayashi J-I, Watanabe J, Miyashita N et al (1981). Evolutionary relationships among five subspecies Mus musculus based on restriction enzyme cleavage patterns of mitochondrial DNA. Genetics 98: 801–816.
Yonekawa H, Moriwaki K, Gotoh O, Miyashita N, Matsushima N, Shi L-M et al (1988). Hybrid origin of Japanese mice ‘Mus musculus molossinus’: evidence from restriction analysis of mitochondrial DNA. Mol Biol Evol 5: 63–78.
Yonekawa H, Tsuda K, Tsuchiya K, Yakimenko LV, Korobitsyna KV, Chelomina GN et al (2003). Genetic diversity, geographic distribution and evolutionary relationships of Mus musculus subspecies based on polymorphisms of mitochondrial DNA. In: Kryukov AP, Yakimenko LV (eds) Problems of Evolution. Dalnauka: Vladivostok. Vol V, pp 90–108.
Acknowledgements
We wish to express our appreciation to MT Kimura, N Takagi, MA Iwasa and T Shiroishi for their valuable advice in this study. We are most grateful to K Aplin for his helpful discussions and comments. We also thank N Aragaki, S-H Han, T Jogahara, AP Kryukov, I Munechika, B Sakumoto, K Sasaki, MH Sinaga and S Suzuki for cooperation in collecting the animals. This study was conducted as part of the Core University Program between Hokkaido University and the Research Centre for Biology, at the Indonesian Institute of Sciences (LIPI) under the title ‘Environmental Conservation and Land Use Management of Wetland Ecosystems in Southeast Asia’, which is supported by the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Terashima, M., Furusawa, S., Hanzawa, N. et al. Phylogeographic origin of Hokkaido house mice (Mus musculus) as indicated by genetic markers with maternal, paternal and biparental inheritance. Heredity 96, 128–138 (2006). https://doi.org/10.1038/sj.hdy.6800761
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800761
Keywords
This article is cited by
-
A boundary between occurrences of mitochondrial cytochrome b haplotypes as well as mito-nuclear discordance in the house mouse Mus musculus from the northern area of Tochigi Prefecture, Japan
Mammalian Biology (2023)
-
House mouse Mus musculus dispersal in East Eurasia inferred from 98 newly determined complete mitochondrial genome sequences
Heredity (2021)
-
What can the geographic distribution of mtDNA haplotypes tell us about the invasion of New Zealand by house mice Mus musculus?
Biological Invasions (2016)
-
Tracing the eastward dispersal of the house mouse, Mus musculus
Genes and Environment (2015)
-
Evolutionary and dispersal history of Eurasian house mice Mus musculus clarified by more extensive geographic sampling of mitochondrial DNA
Heredity (2013)
