
Abstract
Phase polyphenisms are usually thought to reflect plastic responses of species, independent of genetic differences; however, phase differences could correlate with genetic differentiation for various reasons. Mormon crickets appear to occur in two phases that differ in morphology and behaviour. Solitary individuals are cryptic and sedentary whereas gregarious individuals form bands, migrate, and are aposematically coloured. These traits have been thought to be phenotypically plastic and induced by environmental conditions. However, there has been no previous investigation of the extent of genetic differences between solitary and gregarious populations of this widespread North American species. We sequenced two mitochondrial genes, COII and COIII, in samples of Mormon crickets from gregarious populations west of the continental divide and solitary mountain populations primarily east of the divide. Sequencing revealed two genetically distinct clades that broadly correspond with the solitary eastern populations and the mainly gregarious western populations. We used coalescent modelling to test the hypothesis that the species consists of two deep genetic clades, as opposed to a series of equally distinct populations. Results allowed us to reject the null hypothesis that a radiation independent of phase produced these clades, and molecular clock estimates indicate the time of divergence to be approximately 2 million years ago. This work establishes that the solitary populations found in the mountains on the eastern slope are part of a clade that is genetically distinct from the western populations, which are primarily gregarious, and the implications of this apparent correlation between phase and genetic differentiation are discussed.
Similar content being viewed by others


Introduction
Phenotypically plastic traits that fall into discrete classes but do not reflect simple genetic polymorphism are often referred to as phase polyphenisms (Shapiro, 1976). Solitary and gregarious phase polyphenisms are well known in swarming locusts (Orthoptera: suborder Caelifera), but also have been suggested in certain species of flightless bush crickets (katydids: Tettigoniidae) that can reach extraordinarily high population densities and migrate in bands (Cowan, 1990; Gwynne, 2001). Phase polyphenism can arise in response to differing cues during development such as resource availability or density (Evans and Wheeler, 2001). In the desert locust Schistocerca gregaria, for example, population density mediates phenotypically plastic changes in juveniles (Sword and Simpson, 2000; Simpson et al, 2001; Sword, 2002). Under high-density regimes, the locusts develop bright colouration patterns and form gregarious swarms. Such density-dependent phase polyphenisms may have evolved as adaptations to avoid predation pressures in desert locusts, with gregarisation enhancing the beneficial effects of aposematic colouration (Sword, 2002).
Selection on the extent to which phenotypic variation is determined by environmental cues can give rise to genetic differentiation. Although plastic traits themselves may reflect no underlying genetic differences, the degree of plasticity is genetically determined, can vary, and is therefore subject to selection. When selection favours increased plasticity, plastic traits show more variable responses to their environmental cues. Phenotypic variation can thereby influence genetic variation through genetic assimilation in a series of gradual shifts in the trait optima of plasticity (Waddington, 1953; Pál and Miklós, 1999). Recent research has primarily focused on the environmental cues that induce phase polyphenisms in insects such as desert locusts; however, it is also important to study the genetic structure of such populations because of the potential influence that environmentally induced phase polyphenisms can have on genetic divergence.
Alternatively, phase polyphenisms could be genetically distinct if they result from or correlate with different evolutionary histories. For example, many insects show a dimorphism resulting from a trade-off between flight and reproduction (Dingle, 1985). The grasshopper Chorthippus parallelus is normally brachypterous but occasional macropterous forms are found. Two subspecies of this grasshopper differ substantially in how readily macropterous forms appear (Ritchie et al, 1987), perhaps as a result of ecological differences in their evolutionary history. Hence, there will be a statistical association between morph and genetic differentiation that reflects history rather than a more direct association.
Mormon crickets (Anabrus simplex, subfamily Tettigoniinae) have a large geographic range covering nearly the whole of the western US (Gurney, 1939). The species appears to display two distinct phases (Cowan, 1990) that have been assumed to be phenotypically plastic (eg, Gwynne, 1984), but any potential genetic differentiation is unknown. Solitary individuals occur at relatively low densities and are cryptically coloured. They commonly inhabit mountain meadows and have been studied mainly in canyons on the eastern slope of the Rocky Mountains in Colorado (Ueckert and Hansen, 1970; Gwynne, 1984; Lorch and Gwynne, 2000). Gregarious populations occur in various habitats in western North America and are larger and darker in colour than solitary individuals on the eastern slope (Gwynne, 1984). Gregarious Mormon crickets undergo destructive mass migrations and are a major crop pest that can cause tens of millions of dollars in damage in one season (Dunkle, 2001). There is also evidence of life history differences between solitary and gregarious populations (Bailey and Gwynne, pers. obs., 2004).
The phases of Mormon crickets also differ in mating behaviour. Gregarious females observed in north western Colorado compete aggressively for matings, whereas males are choosy of mates. In contrast, solitary crickets on the eastern slope of the Rockies do not show this role-reversal in behaviour (Gwynne, 1981, 1984). Field experiments (Gwynne, 1993) revealed that at high densities, hungry gregarious females are sexually competitive in order to obtain a nuptial gift, the proteinaceous spermatophylax that forms part of the tettigoniid males' spermatophore (Gwynne, 2001).
These studies, in line with taxonomic work (Gurney, 1939), assumed that solitary and gregarious Mormon crickets throughout the range comprise a single widespread species. However, distinct differences in size, colouration, behaviour and possibly life history, as well as the fact that solitary and gregarious populations in Utah and Colorado are usually separated by the North American continental divide, raises the possibility that the two forms have distinct evolutionary histories. Although solitary Mormon crickets can be found at high altitudes, the continental divide may be a significant impediment to gene flow in this flightless species, particularly for solitary individuals because they move less than gregarious individuals (Lorch and Gwynne, 2000). Are there genetic differences between gregarious and solitary populations of A. simplex, or are the distinct differences in phenotype simply a phase polyphenism due to phenotypic plasticity? If there is genetic differentiation, how great is it?
To answer these questions, we sequenced two mitochondrial genes in Mormon crickets, focusing on individuals from gregarious populations close to previous study sites in Utah and north western Colorado and several solitary populations from the eastern slope of the Rockies. However, we also included two solitary populations found west of the continental divide, one of which was about 50 km from gregarious populations. We tested the null hypotheses that (a) the gregarious and solitary populations in previous study sites show no genetic differentiation, and (b) genetic differences between populations within each phase are of equal magnitude. Phylogenetic analyses enabled us to reject both hypotheses. Moreover, the analysis uncovered two very distinct clades of Mormon crickets from the eastern and western slopes of the Rockies that broadly correspond to solitary and gregarious phase.
The population structure in Mormon crickets was further investigated using a nonphylogenetic method of inference. Coalescent modelling is a hypothesis-driven technique that can be used to test alternative evolutionary histories in organisms. Lineages are sampled backwards through a constructed genealogy of the populations in question, and hypotheses are tested by assessing whether the rate at which lineages coalesce deviates significantly from the rate predicted by evolutionary models (Rosenburg and Nordborg, 2002). This technique has been used to address similar questions in several studies, including one of the montane grasshopper Melanoplus oregonensis found in the Rocky Mountains (Knowles, 2001). Coalescent modelling can overcome problems with phylogeographic methods, mainly that the gene history derived from molecular markers may not match the organismal history, which is more likely to occur when examining closely related species or populations (Maddison, 1997; Rosenburg and Nordborg, 2002; Hey and Machado, 2003). It is therefore a superior tool for analysing population structure: we tested the external hypothesis that the phylogenetic pattern observed is consistent with a deep subdivision broadly coincident with the solitary and gregarious forms, for example one that may have occurred during repeated glacial cycles, as opposed to population diversification from a common ancestor independent of phase.
Materials and methods
Sampling
Samples were collected in 2001 from 10 locations in Utah and Colorado, with seven solitary and three gregarious populations represented (Figure 1). One of the solitary populations was collected from Little Brush Creek, Utah, which was 47 km from the gregarious Echo Park and Dinosaur Park populations. The five populations collected on the eastern side of the continental divide were solitary, whereas three of the five populations west of the divide were gregarious.

Map of populations sampled. Gregarious populations are indicated with squares, solitary ones with circles. All three gregarious populations sampled were west of the continental divide, whereas two solitary populations were west of the divide and five east of the divide.
DNA extraction, amplification and sequencing
DNA was extracted following the PureGene protocol (Gentra Systems, Minneapolis, Minn.), and quantified against known concentration standards. Forward and reverse primers C2-J-3279 and TD-N-3862, respectively, were used to amplify a 440 BP region of cytochrome oxidase II mitochondrial gene (COII), and forward and reverse primers C3-J-5014 and C3-N-5460, respectively, were used to amplify a 410 BP region of cytochrome oxidase III mitochondrial gene (COIII). Primer sequences were from Simon et al (1994). PCR reactions were carried out in 50 μl volumes and contained: 0.2 mM each dNTP, 0.20 μM each primer, 1.5 μl 10 × NH4 reaction buffer, 1.5 mM MgCl2, 0.33 μl Biotaq DNA polymerase and 10–300 ng DNA template. The PCR profile for all reactions was: (1) 5 min at 92°C initial denaturation, (2) 10 s at 92°C denaturation, (3) 15 s at the annealing temperature (47°C for COII and 46°C for COIII), (4) 1 min at 72°C extension, (5) 39 repeats of steps 2–4 and (6) 10 min at 72°C final extension. PCR products were checked on 1.5% agarose gels and sequenced using a Beckman–Coulter automated sequencer. Raw sequences were edited and aligned using Chromas 2.12 (Technelysium Pty Ltd, 2001) and ClustalX 1.81 (Thompson et al, 1997), respectively. Forward and reverse sequences were produced for each individual and aligned to reduce the likelihood of sequencing errors. Generally there was little discrepancy, although sequences were rerun if forwards and reverses did not correspond. All sequences corresponded in the end.
Phylogeographic analysis
We tried but were unable to sequence both COII and COIII in various outgroup species because of difficulties with PCR amplification. We have therefore included two tettigoniid species as outgroups: Ephippiger ephippiger (subfamily Bradyporinae) and Kawanaphila gidya (Zaprochilinae). Outgroups in the analysis were sequenced for either COII or COIII.
We used Modeltest 3.06 (Posada and Crandall, 1998), in conjunction with PAUP* 4.0 (Swofford, 1998), to assess which model of substitution was most suitable for these sequences, and the Tamura-Nei model with a gamma distribution shape parameter of 0.0937 was most appropriate. Instead of analysing sequences from both genes separately, we used the partition homogeneity test in PAUP* to test if the phylogenetic signals from the two genes were congruent. They were, so the two sequences were concatenated and analysed together. To align the outgroups with the concatenated sequences, we simply coded the absent gene as missing data. A maximum likelihood tree for the concatenated sequences was constructed in PAUP* (Swofford, 1998) using 20 replicates. A neighbour-joining tree with bootstrap values generated using 500 replicates was also produced in MEGA v.2.1 (Kumar et al, 2001) and compared with the maximum likelihood tree.
We divided our subsequent analyses into two categories: the first set was to test external hypotheses about whether genetic differentiation in our populations of Mormon crickets is coincident with phase, so these analyses were carried out independent of the structure found in the mtDNA phylogeny. The second set was carried out to generate summary statistics on the genetic clades uncovered in the phylogeny. These analyses therefore specifically addressed the structure found in the mtDNA phylogeny and were carried out independent of phase.
Coalescent analysis
Coalescent modelling can be used to evaluate the level of discordance between population histories and gene trees. One measure of this is Slatkin's s, which measures the number of additional parsimony steps in a given tree that are needed for the gene tree to correspond to the population history – denoted as a population tree – in which it is contained (Slatkin and Maddison, 1989; Knowles, 2001). Therefore, instances of incomplete lineage sorting, called ‘deep coalescences’, cause greater discord between gene and population trees, which produces a higher s value.
Our null hypothesis was that the population structure observed in Mormon crickets results from population fragmentation independent of the phases, which would predict no significant genetic differentiation between populations grouped by phase. The alternative hypothesis is that the two forms reflect distinct evolutionary histories resulting from an older division within the species, for example due to glacial periods in the Pleistocene.
We used the program Mesquite v.1.0 (Maddison and Maddison, 2004) to test these two evolutionary histories. First, we created a star-shaped population tree with 10 branches that reflects the evolutionary history under the null hypothesis – a fragmentation independent of phase (Figure 2). Each branch represents one population that we sampled. We then simulated 1000 gene trees within this population tree using neutral coalescence. To test the alternative hypothesis, we contained the simulated gene trees within a V-shaped population tree that had two deep branches representing a split between solitary and gregarious populations – the ‘deep clade’ tree (Figure 3). We used Slatkin's s to measure the discordance between the gene trees simulated in the null population tree and the alternative deep clade tree in which they were contained. Finally, we contained our reconstructed empirical gene tree produced in PAUP* within the deep clade tree and measured the discordance. If the s value of our reconstructed tree falls below 95% of the distribution of simulated gene tree s values, we can reject the null model in favour of the deep clade model. It should be emphasised that we did not test the relative timing of population divergence under these scenarios; here, we concentrate on only testing the particular hypothesis that there is a single large genetic subdivision broadly coincident with the phases of this species.

Diagram of null population model. Lines within the tree represent individual gene trees, or lineages, and each branch represents one population (labels correspond to Figure 1). This star-shaped tree models rapid fragmentation of an ancestor, with populations diverging independent of phase. We simulated 1000 gene trees within this population tree using neutral coalescence.

Diagram of ‘deep clade’ alternative population model in which simulated and empirical gene trees were contained. The two branches represent a deep phylogeographic split corresponding with phase.
Separately, an overall nested AMOVA (populations nested within phase) was performed in Arlequin ver. 2.000 (Schneider et al, 2000) to assess the proportion of genetic variation that was distributed between the phases and among populations within phases, which might be expected to differ given the differences in movement.
Summary statistics
Two analyses were performed to generate summary statistics on the two clades uncovered by our phylogenetic analysis. We used the DNADIST v. 3.6 executable in PHYLIP (Felsenstein, 2004) to determine pairwise distance measures using a Jukes–Cantor correction between individuals in the two clades, from which we then calculated the mean percentage of sequence divergence between the clades following the method of Lunt et al (1998). We extrapolated an approximate time since divergence from this information. To gauge differences in population structure within the two clades, a separate AMOVA was performed on each to compute the percentage of among vs within population variation, allowing us to assess whether differentiation among populations was greater within either clade. AMOVAs were perfomed in Arlequin ver. 2.000 (Schneider et al, 2000).
Results
Phylogeographic analysis
The concatenated sequences from 37 individuals yielded an 850 BP fragment with 79 parsimony-informative characters and 20 discrete haplotypes. The topology of the maximum likelihood tree was similar to that of a neighbour-joining tree constructed in MEGA 2.1 (Kumar et al, 2001) using the Kimura–Nei model.
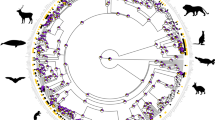
There are two noteworthy patterns in the tree, shown in Figure 4. First, the samples fall out into two discrete clades that broadly correspond with phase: a central and eastern Colorado mountains clade of only solitary populations (‘eastern’ clade) and a clade of populations from west of the Colorado Rockies that is mainly comprised of gregarious populations (‘western’ clade). The Little Brush Creek solitary population (coded ‘B’) is the only solitary population that groups with the gregarious onesin the western clade. Despite this exception, an overall nested AMOVA indicated significant genetic differentiation between solitary and gregarious populations with 10.57% of the genetic variation distributed between the phases (P=0.028±0.005). Second, compared to the western clade, there is more genetic structure in the eastern clade, where many populations form monophyletic groups and are supported by high bootstrap values. Branch lengths in the western clade are shorter, populations generally do not form monophyletic groups, and bootstrap support is low.

Tamura-Nei NJ tree using COII and COIII sequences combined (MEGA v.2.1). Bootstrap values over 80% are indicated. Sample labels are as in Figure 1, and western and eastern clades are indicated. Little Brush Creek solitary samples falling in the western clade with gregarious populations are labelled with asterisks (*).
Coalescent analysis
Figure 5 shows the s values produced when we took 1000 gene trees simulated by neutral coalescence in our star-shaped null population history model and contained them within the V-shaped deep clade model. The s value of our reconstructed empirical tree when contained within the alternative model was 3, indicated by the dashed line in the histogram. It falls below 95% of the distribution of simulated gene trees, so our empirical tree rejects the null hypothesis in favour of the alternative deep clade model.

Histogram of s-values generated when trees constructed using neutral coalescence in the null population model were contained within the alternative deep clade model. The s-value generated when the mtDNA gene tree for our Mormon cricket populations was contained within the alternative deep clade model is indicated by the dashed line.
Divergence of geographic clades
The mean percentage of sequence divergence between individuals from different clades was 3.98, SD ±0.43%. Given an approximate rate of sequence divergence of 2% per million years (Brown et al, 1979; Lunt et al, 1998; Grant and Miles, 2002), divergence time for these clades is between approximately 1.78 and 2.21 million years ago.
Separate AMOVAs for each clade indicate that there is more differentiation within the eastern clade, with 80.86% of genetic variation attributable to between population differences, whereas in the western clade only 39.07% of the variation is between populations. Conversely, within population variation is lower in the eastern clade (19.14%) than in the western clade (60.93%).
Discussion
The broad genetic division between western (mainly gregarious) and eastern (solitary) populations of A. simplex is an unexpected result given the previous assumption that gregariousness in this species is a phenotypically plastic trait (Gwynne, 1993). Our results show that there is a significant genetic division within A. simplex; populations on the east and west slope of the Rockies form two discrete clades. We raised two contrasting hypotheses regarding the nature of these clades and the phase of the populations comprising them. In one, populations differ without respect to phase whereas in the other, the phases represent or coincide with a deep phylogeographic split within the species. Under the former scenario, we would have expected a phylogenetic analysis to indicate either complete panmixis or paraphyletic groups for each population over the whole species. However, our analyses reject the former scenario in favour of the latter; in the populations that we sampled, there is significant genetic differentiation between the phases and the phylogeographic history more strongly supports the second hypothesis. The division is not perfect, however, because the solitary Little Brush Creek population – sampled just 50 km from gregarious populations in the west – falls in the clade with all gregarious populations. Given that the data overall support a complex history behind the phases, incorporating plasticity, what does the genetic division suggest about the evolutionary history of the two forms?
Early origin of the division
Based on the sequence divergence between the eastern and western clade, we estimate that the major clades diverged around 2 million years ago. Having diverged before the last ice age, they must have quite distinct evolutionary histories during which genetic differences have accumulated. Such differences may influence a range of traits, including the environmentally induced threshold influencing the switch point between alterative phases. In general, such switch points between alternative morphologies are known to be genetically determined and to evolve, sometimes rapidly (eg Tomkins and Brown, 2004). An alternative to our favoured hypothesis of differing thresholds between two genetically distinct forms is that the threshold remains similar but environmental conditions are ‘fixed’ between eastern and western populations. In addition to the genetic divergence, these clades differ in morphology and life history characteristics.
There is no formal agreed definition for subspecies (like species), but a working hypothesis is that subspecies should differ at numerous concordant traits yet lack reproductive isolation (Avise and Ball, 1990). The continental divide is a site of numerous hybrid zones between subspecies (though see Swenson and Howard, 2004). Our analyses indicate that the two clades within Mormon crickets have probably been evolving independently for around 2 million years, which is consistent with other studies of subspecific variation (eg, Hewitt, 1996; Lunt et al, 1998) and must have occurred over several glacial cycles. Indeed, many sister species of North American birds show less mtDNA divergence (Lovette and Bermingham, 1999; Lovette, 2005). During periods of allopatry, genetic differences between eastern and western Mormon crickets could have accumulated either by genetic drift or via selection for variation in response to differing environmental cues. Further studies of Mormon crickets will be necessary to fully assess the level of divergence between these forms and over what geographic scale the division remains correlated with phase. A partial Mantel analysis of the data shows that, after controlling for isolation by distance, classification of samples as east or west of the continental divide is an almost significant predictor of genetic distance (P=0.056), whereas solitary vs gregarious classification is not. It is therefore likely that the genetic division results primarily from glacial refugia and the association with phase is less direct, reflecting the possible influence of plasticity in conjunction with phylogeographic history. Current studies are addressing this with more extensive geographic sampling and analyses of additional morphological and behavioural traits.
In our current samples, there is one interesting and important exception to the broad coincidence between phases and the deepest clade: the solitary samples from Little Brush Creek, UT cluster in the western clade. There are several possible explanations for this. Despite the general coincidence, exceptions are expected given the importance of environmental condition in any one year in generating a phase switch. This population might experience unusual local conditions, or have a particularly high switch point for becoming gregarious. Based on food-manipulation experiments, Gwynne (1993) concluded that courtship role reversal, seen in gregarious populations, is a result of low food availability due to high population density. Further support for the idea that food quality causes a switch in phase from solitary to gregarious comes from other studies of Mormon crickets and the desert locust Schistocerca gregaria (Gwynne, 1984; Simpson et al, 2001).
Other possibilities include migration and or hybridisation. Gregarious bands of Mormon crickets originating in the west have been observed within 1 km of the Little Brush Creek site (Bailey, pers. obs., 2004) and if gregarious individuals strayed from their band and colonized the area, subsequent generations may have matured in conditions that were not conducive to gregarious band formation, for example, low density, high nutritional resources, etc., and become solitary. This could explain why there is virtually no genetic difference between the Little Brush Creek solitary population and all the gregarious populations. Alternatively, there may be a high level of gene flow into the Little Brush Creek population because gregarious Mormon crickets are by their nature very mobile. If no barriers to reproductive isolation exist between the two phases, bands of gregarious individuals from nearby may have interbred with Little Brush Creek crickets in the past. Although solitary Mormon crickets at Little Brush Creek reach reproductive maturity 4–6 weeks later than the gregarious ones nearby (Bailey and Gwynne, pers. obs., 2004), once they are adults they may be capable of mating with migratory gregarious individuals later in the season.
Differences in population structure between eastern and western clades
Separate AMOVA analyses indicate that populations in the western clade show much less genetic structure than populations in the eastern clade. This is not surprising because gregarious Mormon crickets form bands that travel far and wide, sometimes marching further than 1 km per day (Lorch and Gwynne, 2000). There are even accounts of separate bands intersecting one another (MacVean, 1987), and these band movements will provide opportunities for interpopulation matings and gene flow. Populations in the eastern clade, however, are much less likely to disperse because they inhabit small areas of meadow habitat within mountain canyons and do not move over very long distances (Lorch and Gwynne, 2000), thereby decreasing the likelihood of gene flow between other solitary populations. Over historical timescales, these different migration rates may have had a homogenizing effect on the genetic structure of gregarious populations and may have facilitated differentiation among solitary populations.
Implications
The specific assumption in Gwynne's (1984) observational work showing different sex roles in Mormon crickets was that eastern Poudre Canyon and western Dinosaur National Monument populations were not genetically different. The mtDNA evidence presented here, however, challenges the implicit assumption that phase polyphenisms, and hence sex role-reversal, in those populations are solely plastic traits, in favour of the idea that phase broadly coincides with an ancient subdivision within the species. As with any example of phenotypic plasticity, exceptions will occur and the Little Brush Creek population provides an example. Further studies must concentrate on improving the geographic sampling to better assess the extent of coincidence between phase and genetic differentiation.
Even though the genetic differentiation seen in Mormon crickets is consistent with subspecies or even species status, such a conclusion should not be based simply on levels of genetic differentiation. What makes this system novel is that a suite of morphological, behavioural and life history traits are broadly coincident with the solitary/gregarious phase difference in addition to the genetic differentiation. If we are correct in considering the differentiation between clades similar to that between subspecies, it will be worth re-examining other trait differentiation between the clades, especially including behaviours and other traits with the potential to influence reproductive isolation and gene flow between these forms.
References
Avise JC, Ball RJM (1990). Principles of genealogical concordance in species concepts and biological taxonomy. In: Futuyma D, Antonovics J (eds) Oxford Surveys in Evolutionary Biology. Oxford University Press: Oxford, UK.
Brown WM, George M, Wilson AC (1979). Rapid evolution of animal mitochondrial DNA. Proc Natl Acade Sci USA 76: 1967–1971.
Cowan FT (1990). The Mormon cricket story, Montana State University, Agric. Exp. Station Special Report #31: 7–42.
Dingle H (1985). Migration. In: Kerkut GA, Gilbert CI (eds) Insect physiology, biochemistry and pharmacology, Vol. 9. Behaviour. Pergamon press: Oxford, UK.
Dunkle R (2001). Testimony before the subcommittee on national parks, recreation, and public lands, committee of natural resources, United States house of representatives http://resourcescommittee.house.gov/archives/107cong/parks/2001july19/dunkle.htm.
Evans JD, Wheeler DE (2001). Gene expression and the evolution of insect polyphenisms. BioEssays 23: 62–68.
Felsenstein J (2004). PHYLIP (Phylogeny Inference Package), Version 3.6b. University of Washington: Seattle, WA.
Grant MW, Miles MA (2002). An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol Biol Evol 19: 748–761.
Gurney AB (1939). Aids to the identification of the Mormon and Coulee crickets and their allies (Orthoptera: Tettigoniidae, Gryllacrididae). United States Department of Agriculture Bureau of Entomology and Plant Quarantine. June.
Gwynne DT (1981). Sexual difference theory: Mormon crickets show reversal in mate choice. Science 213: 779–780.
Gwynne DT (1984). Sexual selection and sexual differences in Mormon crickets (Orthoptera: Tettigoniidae, Anabrus simplex). Evolution 38: 1011–1022.
Gwynne DT (1993). Food quality controls sexual selection in Mormon crickets by altering male mating investment. Ecology 74: 1406–1413.
Gwynne DT (2001). Katydids and Bush-Crickets. Comstock Publishing Associates: Ithaca, NY.
Hewitt GM (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58: 247–276.
Hey J, Machado CA (2003). The study of structured populations – new hope for a difficult and divided science. Nature 4: 535–543.
Knowles LL (2001). Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshoppers. Mol Ecol 10: 691–701.
Kumar S, Tamura K, Jakobsen IB, Nei M (2001). MEGA2: Molecular Evolutionary Genetics Analysis. Arizona State University: Tempe, Arizona, USA.
Lorch PD, Gwynne DT (2000). Radio-telemetric evidence of migration in the gregarious but not the solitary morph of the Mormon cricket (Anabrus simplex: Orthoptera: Tettigoniidae). Naturwissenschaften 87: 370–372.
Lovette IJ (2005). Glacial cycles and the tempo of avian speciation. TREE 20: 57–59.
Lovette IJ, Bermingham E (1999). Explosive speciation in the New World Dendroica warblers. Proc R Soc Lond B 266: 1629–1636.
Lunt DH, Ibrahim KM, Hewitt GM (1998). MtDNA phylogeography and postglacial patterns of subdivision in the meadow grasshopper Chorthippus parallelus. Heredity 80: 633–641.
MacVean CM (1987). Ecology and management of the Mormon cricket, Anabrus simplex Haldeman. In: Capinera JL (ed) Integrated Pest Management on Rangeland: a Shortgrass Prairie Perspective. Westview Press: Boulder, Colo. pp 116–136.
Maddison WP (1997). Gene trees in species trees. Syst Biol 46: 523–536.
Maddison WP, Maddison DR (2004). Mesquite: a modular programming system for evolutionary analysis v.1.0. http://mesquite-project.org.
Pál C, Miklós I (1999). Epigenetic inheritance, genetic assimilation and speciation. J Theor Biol 200: 19–37.
Posada D, Crandall KA (1998). Modeltest: testing the model of DNA substitution. Bioinfomatics 14: 817–818.
Ritchie MG, Butlin RK, Hewitt GM (1987). Causation, fitness effects and morphology of macropterism in Chorthippus parallelus (Orthoptera: Acrididae). Ecol Entomol 12: 209–218.
Rosenburg NA, Nordborg M (2002). Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nature 3: 380–390.
Schneider S, Roessli D, Excoffier L (2000). Arlequin ver. 2.000: A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva: Switzerland.
Shapiro AM (1976). Seasonal Polyphenism. Evol Biol 9: 259–333.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87: 651–701.
Simpson SJ, Despland E, Hägele BF, Dodgson T (2001). Gregarious behavior in desert locusts is evoked by touching their back legs. Proc Natl Acad Sci USA 98: 3895–3897.
Slatkin M, Maddison WP (1989). A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123: 603–613.
Swenson NG, Howard DJ (2004). Do suture zones exist? Evolution 58: 2391–2397.
Swofford DL (1998). PAUP*: Phylogenetic Analysis Using Parsimony and other methods. Beta version 4.0b10. Sinauer Associates: Sunderland, Mass.
Sword GA (2002). A role for phenotypic plasticity in the evolution of aposematism. Proc R Soc Lond B 269: 1639–1644.
Sword GA, Simpson SJ (2000). Is there an intraspecific role for density-dependent colour change in the desert locust? Anim Behav 59: 861–870.
Technelysium Pty. Ltd (2001) Chromas 2.12. Copyright 1998–2001. PO Box 247, Helensvale, Queensland 4212, Australia.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997). The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24: 4876–4882.
Tomkins JL, Brown GS (2004). Population density drives the local evolution of a threshold dimorphism. Nature 431: 1099–1103.
Ueckert DN, Hansen RN (1970). Seasonal dry-weight composition in diets of Mormon crickets. J Econ Ecol 63: 96–98.
Waddington CH (1953). Genetic assimilation of an acquired character. Evolution 7: 118–126.
Acknowledgements
We thank P Lorch for helping in collecting crickets, L Hockham for his valuable assistance in the early stages of molecular lab work, K Vahed for providing outgroup samples, R Hamill for encouragement during those long dark hours in the lab and D Kidd and L Knowles for elucidating the coalescent. This work was funded by an Overseas Research Students Award Scheme and the University of St Andrews.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bailey, N., Gwynne, D. & Ritchie, M. Are solitary and gregarious Mormon crickets (Anabrus simplex, Orthoptera, Tettigoniidae) genetically distinct?. Heredity 95, 166–173 (2005). https://doi.org/10.1038/sj.hdy.6800704
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800704
