
Abstract
A horizontal transmission of a geminiviral DNA sequence, into the germ line of an ancestral Nicotiana, gave rise to multiple repeats of geminivirus-related DNA, GRD, in the genome. We follow GRD evolution in Nicotiana tabacum (tobacco), an allotetraploid, and its diploid relatives, and show GRDs are derived from begomoviruses. GRDs occur in two families: the GRD5 family's ancestor integrated into the common ancestor of three diploid species, Nicotiana kawakamii, Nicotiana tomentosa and Nicotiana tomentosiformis, on homeologous group 4 chromosomes. The GRD3 family was acquired more recently on chromosome 2 in a lineage of N. tomentosiformis, the paternal ancestor of tobacco. Both GRD families include individual members that are methylated and diverged. Using relative rates of synonymous and nonsynonymous nucleotide substitutions, we tested for evidence of selection on GRD units and found none within the GRD3 and GRD5 families. However, the substitutions between GRD3 and GRD5 do show a significant excess of synonymous changes, suggesting purifying selection and hence a period of autonomous evolution between GRD3 and GRD5 integration. We observe in the GRD3 family, features of Helitrons, a major new class of putative rolling-circle replicating eukaryotic transposon, not found in the GRD5 family or geminiviruses. We speculate that the second integration event, resulting in the GRD3 family, involved a free-living geminivirus, a Helitron and perhaps also GRD5. Thus our data point towards recurrent dynamic interplay between geminivirus and plant DNA in evolution.
Similar content being viewed by others

Introduction
Rolling-circle replication
The IS91 family of bacterial elements transpose via rolling-circle (RC) replication (RCR), first characterised in single-stranded (ss) DNA bacteriophages, is also carried out by bacterial plasmids during conjugative transfer and in some cases during vegetative replication. A critical step in RCR is mediated by the replication protein (Rep), which is a sequence- and strand-specific endonuclease/ATPase/ligase. In RCR, Rep generates ssDNA circular monomers from a cis-essential origin (ori) on a double-stranded (ds) replicative form (RF) (Hanley-Bowdoin et al, 2000). Kapitonov and Jurka (2001) identified a major new class of putative RCR transposon in eukaryotes. They are called Helitrons and make up a significant proportion of some, perhaps most, plant and animal genomes, notably 2% of the genomes of Arabidopsis thaliana and Caenorhabdites elegans.
In eukaryotes, various ssDNA viruses also replicate by RCR. These include in plants, the geminiviruses and the more recently discovered nanoviruses, and in animals the circoviruses. All share sequence and structural features with prokaryotic RC replicators and their Rep is evolutionarily related, leading to speculation that these viruses evolved from prokaryotic ssDNA replicons (Koonin and Ilyina, 1992; Gibbs and Weiller, 1999). Kapitonov and Jurka (2001) suggest that Helitrons are the missing evolutionary link between prokaryotic RC elements and geminiviruses.
Geminivirus-related DNA sequences in Nicotiana genomes
Previously we discovered a family of repetitive plant DNA sequences, thought to have arisen via illegitimate integration of geminiviral DNA, hence geminivirus-related DNA (GRD), into the nuclear genome of an ancestor to some species in the plant genus Nicotiana (Day et al, 1991; Bejarano and Lichtenstein, 1994; Bejarano et al, 1996; Ashby et al, 1997). GRD contains a degenerate and truncated geminiviral rep gene (also known as AL1 and AC1) plus the untranscribed intergenic region carrying the origin of replication (ori).
Tobacco, an allotetraploid between ancestors of Nicotiana sylvestris and Nicotiana tomentosiformis (Lim et al, 2000), acquired GRD from the N. tomentosiformis parent (Murad et al, 2002). GRDs are also found in two close relatives of N. tomentosiformis, namely Nicotiana tomentosa and Nicotiana kawakamii (Ashby et al, 1997) where they occur in tandem array at homoeologous loci (Lim et al, 2000).
We present new sequence data and phylogenetic analyses to establish relationships between the different GRD units and between GRD and related geminiviruses. We show that GRD has structural features in common with Helitrons and these features are not present in geminiviruses. We also make use of the chronology of Nicotiana speciation and allopolyploid formation to provide historical landmarks, which help establish the nature of GRD integration and its diversification. We compare patterns of DNA substitution in GRD and Nicotiana to infer the historical pattern of selection and mutational bias that have acted on the GRD sequences.
Material and methods
Sequence data
Nucleotide sequences for geminivirus replication-associated proteins (Rep) were obtained from GenBank (Table 1). The 21 geminivirus-related DNA (GRD) sequences, in addition to a range of rep sequences, from representatives of three genera of geminiviruses are presented in Table 1. GRD sequences were obtained either from GenBank (Ashby et al, 1997) or by direct DNA sequencing (Table 1). Ashby et al (1997) show alignments of GRD sequences against begomovirus rep. The 21 GRD sequences included representatives of four previously identified types: GRD2, GRD3, GRD5 and GRD53 (Ashby et al, 1997). Alignments were initially obtained by using the Clustal program (Higgins and Sharp, 1988) and edited manually using the GeneDoc program to retain an ORF in rep; frame-shift mutations in GRD suggest that it does not encode any functional proteins.
Relationships among GRD and geminivirus sequences
Relationships between GRD and geminviral sequences were inferred by phylogenetic analysis of nucleotide sequences. Four representative GRD sequences (GRD2NT, GRD3, GRD53 and GRD5NT1) were sampled for the purpose of analysing their relationship to known geminivirus rep sequences. Based on this analysis, two closely related geminivirus sequences were identified for use as outgroups in a phylogenetic analysis of all available GRDs. Both analyses were based on the maximum likelihood (ML) criterion as implemented in PAUP* (Swofford, 1993). Analysis assumed the Hasegawa–Kishino–Yano substitution matrix (HKY85 model; Hasegawa et al, 1985) combined with the discrete gamma model of rate variation (Yang, 1994). Nonparametric bootstrapping was used to assess relative support for individual nodes (Felsenstein, 1985; Penny and Hendy, 1985).
Detecting the action of selection
We used the phylogenetic framework of Goldman and Yang (1994) to estimate ω, where ω=dN/dS, and dN and dS are nonsynonymous (causing amino-acid replacement) and synonymous (silent) substitution rates, respectively. The analyses were based on an ML model of codon substitution as implemented in the ‘codoml’ program of the PAML package (Yang, 1997). We employed a correction for transition/transversion rate bias and codon usage bias, as these features of DNA sequence evolution can have a significant effect on the estimation of dN and dS (Bielawski et al, 2000; Yang and Nielsen, 2000; Dunn et al, 2001). To test the hypothesis of selective neutrality, models assuming neutrality (Model A: constrained so that ω=1) were compared to a model with no such constraint (Model B: ω estimated as a free parameter) by using a likelihood ratio test (Yang and Bielawski, 2000).
Results
Phylogenetic analysis of rep and GRD
The family Geminiviridae consists of four genera: Mastrevirus, which has monopartite genomes, is transmitted by leafhoppers and mostly infect grasses (monocots); Begomovirus, which has mostly bipartite genomic components (A and B genomes), is transmitted by the whitefly Bemisia tabaci to dicots; Curtovirus, with a monopartite genome, is transmitted by leafhoppers to dicots; and Topocuvirus, which contains only a single virus species, has a monopartite genome and is transmitted by a single species of treehopper to dicots.
Geminivirus replication requires a replication protein encoded by rep which binds to recognition sequence repeats, iterons, to initiate RCR at a stem–loop structure in the intergenic region. Although quite divergent, the rep gene of geminiviruses showed sufficient similarity to GRD to allow alignment; regions of ambiguous alignment were excluded from further analysis. Mean pairwise sequence divergence (±STD) between GRDs and Begomovirus was 36% (±6%), and between GRDs and Mastrevirus was 54% (±3%).
Phylogenetic analysis of geminivirus genera and GRD yielded the topology shown in Figure 1. This tree clearly suggests that GRD sequences shared a common ancestor whose origin was within Begomovirus (Figure 1). The apparent monophyletic origin of these GRD sequences was strongly supported (PB=97), with this GRD clade sister to a clade comprised of BGYMV, ToMoV and CdTV (Figure 1). These findings suggest a single insertion of a Begomovirus, into a Nicotiana ancestor (but see later for an alternative interpretation of this topology). Based on this topology, we selected two lineages (BGYMV and TGMV) to serve as outgroups for a larger phylogenetic analysis of relationships among the GRD sequences.

Phylogenetic analysis of GRD and representative geminivirus rep sequences. The tree demonstrates the relationships of geminivirus genera and suggests that GRD sequences are derived from Begomovirus. The location of the root was inferred from an earlier analysis of geminivirus and nanovirus Rep amino-acid sequences.
Phylogenetic analysis of all 21 GRD sequences available (Table 1) indicated that GRD sequences comprise two distinct clades: (i) the GRD5 family (which includes GRD5 deletion derivatives, GRD5Δ plus GRD2 in N. tomentosa) and (ii) the GRD3 family (which includes GRD3 deletion derivatives, GRD3Δ and GRD53) (Figure 2). Within the GRD5 and GRD3 families, internal branches are very short, suggesting rapid diversification of GRDs within each clade. The units of the GRD3 family do not cluster into groups of N. tomentosiformis and Nicotiana tabacum origin. Thus the divergence among GRD3 units probably occurred before the interspecific hybridisation event that created tobacco (Figure 2) (Murad et al, 2002). Likewise the GRD5 family, which occurs in N. tabacum, N. tomentosiformis and N. kawakamii, shows no clustering of units from the same species (Figure 2). This pattern suggests that the majority of genetic differences between current GRD5 units had already accumulated in the common ancestor of these three species. N. tomentosa lacks full-length GRD5 elements, but carries 5–15 copies of GRD2 (Ashby et al, 1997) at the chromosome 4 locus. These units resemble the GRD5Δ of other species, but branch out separately.

ML tree showing within-group relationships of GRD sequences. GRD nomenclature: GRD sequences identified with NT are from N. tabacum, NK from N. kawakamii, NA from N. tomentosa and NF from N. tomentosiformis. GRD occurs as two distinct subfamilies: GRD3 family (includes clones labelled GRD53) and GRD5 family (includes GRD2).
Selective pressure in rep and GRD
In the geminiviral rep gene, not surprisingly, patterns of sequence divergence at the three codon positions are consistent with the rep gene evolving under purifying selection on the amino-acid product: sequence divergence at first and second positions is lower than at third (degenerate) positions (first=0.20, second=0.17, third=0.30). Since GRD is now noncoding, we would expect similar sequence divergence at all three codon positions. Yet, surprisingly, patterns of sequence divergence, averaged over all GRD families, also showed more divergence at the third codon position (first=0.10, second=0.08, third=0.20).
In protein coding sequences, selection can be more accurately identified by the ratio of nonsynonymous (amino-acid replacement) and synonymous (silent) substitution rates, dN and dS respectively. The difference between these two rates, measured as the ratio ω=dN/dS, reflects the effect of selection on the protein product of the gene (Kimura, 1983). For example, if nonsynonymous mutations are deleterious, then purifying selection will reduce or prevent their fixation rate and dN/dS will be less than 1. However, if nonsynonymous mutations are neutral, then they will be fixed at the same rate as synonymous substitutions and dN/dS will be close to 1.
The ML estimate of the ω ratio was obtained for geminiviral rep (Figure 1, excluding all GRDs) and GRD (Figure 2, excluding all reps). As rep encodes an essential replication protein, it is not surprising that ω is much less than 1 (ω=0.1), indicating evolution by strong purifying selection. Indeed a likelihood ratio test showed that ω is significantly less than the neutral expectation (Table 2). The same pattern was also observed within GRD, with an estimate of ω=0.33, also significantly less than the neutral expectation (Table 2). To investigate if purifying selection has operated throughout the entire history of the GRD clade, we re-evaluated ω independently in the GRD5 and the GRD3 family clades. In contrast, ω is not significantly different from the neutral expectation (GRD5, ω=0.7; GRD3, ω=0.85) within these two clades (Table 2). These findings suggest that there is purifying selection between the two families, but that it is weaker than within rep sequences of geminiviruses, and that there is no evidence for selection within the families (Figure 2). This raises the possibility of two integration events (see Discussion).
Analysis of CG dinucleotide frequencies
There is considerable evidence that methylated CpG dinucleotides represent mutational hot spots due to spontaneous (Bird, 1986; Frederico et al, 1990) or enzymatic (Zingg et al, 1998) deamination to TpG dinucleotides. Since GRD5 is heavily methylated in cytosine at CG and CNG nucleotides (Kovarik et al, 2000), we investigated whether cytosine methylation has resulted in a depressed frequency of CG dinucleotides because of higher rates of C to T transitions over long periods of time (which would generate TG dinuclotides). Analysis of the content of individual dinucleotides in GRD monomers is shown in Figure 3. The CG frequency is more than 50% below expectation (expectation is the frequency of dinucleotides found in aligned geminivirus rep and ori sequences). There is also an accompanying excess of CA and TG dinucleotides (Figure 3). On the other hand, the relative content of individual CNG trinucleotides in GRD, compared to geminivirus rep and ori, revealed no detectable loss of cytosines at these sites (results not shown). There is also an excess of CC and GG dinucleotides, which cannot be explained by methyl C to T transitions.

A comparison of average dinucleotide frequencies in 18 GRD clones (of Ashby et al, 1997) and the average for rep and ori of five geminiviruses (TGMVA, BGMVA, MYMVA, PYMVA and TLCV). The bar chart shows 10 dinucleotides; not all 16 sets of dinucleotides are presented since the frequency of AA, CC, CA, AG, GA and AC (shown) must equal that of TT, GG, TG, CT, TC and GT respectively (not shown). These 10 dinucleotides are plotted against (observed−expected)/expected dinucleotide frequencies. The expected is the random frequency of a given dinucleotide occurring in a given sequence and was scored as follows: suppose the frequency of C's is 30% and of G's is 30% (scoring on both strands), then the expected frequency of CG dinucleotides (and indeed also GC dinucleotides) is 0.3 × 0.3 × nucleotide length.
Helitron-like properties of GRD
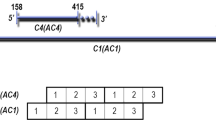
The GRD3 family, but not the GRD5 family of elements, share essential structural components of plant Helitrons: a 3′ palindromic hairpin loop sequence with a downstream CTAG motif (Figure 4). Furthermore, the geminiviral Rep, also present in both GRD3 and GRD5 families, shows some identity to DNA helicases. Thus GRD3 family members have the structural hallmarks of sequences involved in RCR that are also found in a range of nonautonomous Helitrons (Feschotte and Wessler, 2001).

In animals and plants, Helitrons have a conserved 16–20 bp palindrome of DNA making a hairpin loop with a 10–12 bp downstream CTAG sequence (Kapitonov and Jurka, 2001). The GRD3 family of elements, like all rolling-circle replicons, have the stem–loop structure. They also have the conserved downstream CTAG.
Discussion
Evolution of GRD via cytosine methylation
It has been proposed that DNA methylation at cytosine has evolved as a host-defence mechanism to protect the genome against the movement of genomic parasites, such as transposable elements, by repressing their movement through chromatin condensation and/or inhibiting gene expression (Yoder et al, 1997). Geminivirus genomes are not themselves methylated (Brough et al, 1992). In contrast, the genome of N. tabacum has particularly high overall levels of methylation, especially at several tandem repeats including GRD sequences (Kovarik et al, 1997). The comparison of the content of CG and TG in GRD, with that of geminivirus rep and ori, indicates a slight shift in nucleotide composition consistent with elevated rates of C to T transitions in GRD relative to geminiviruses. This pattern implies that the currently observed methylation in GRD is long-standing, perhaps dating back to soon after GRD became integrated into the plant genome.
Phylogenetic reconstructions of GRD families
Phylogenetic reconstructions of GRD clones reveal that there are two distinct GRD families, the GRD3 and GRD5 families. Both families diverge from begomoviruses suggesting they are descendants of geminiviral DNA. Previous studies have shown that both GRD families are found in blocks of a few tens to a hundred tandem direct repeats: the GRD5 family, found in N. tabacum, N. tomentosiformis, N. kawakamii and N. tomentosa (which carries the variant GRD2), occurs on homoeologous chromosome 4 in each species (T4 in tobacco) (Lim et al, 2000); the GRD3 family, found only in a lineage of N. tomentosiformis and in tobacco, occurs on chromosome 2 and T2 respectively (Murad et al, 2002).
The relationship between Nicotiana species in section Tomentosae has been determined by comparing the occurrence and distribution of nine repetitive sequences by fluorescent in situ hybridisation (Lim et al, 2000). The GRD-carrying species appear to have inherited GRD from a common ancestor within Tomentosae (Figure 5), and a lineage of N. tomentosiformis carried GRD3 and GRD5 to tobacco (Murad et al, 2002) (Figure 5). As the phylogenetic analysis of GRD clones (Figure 2) reveals that there is no species-specific clustering of GRD units for either the GRD5 or GRD3 family, this indicates that after integration of a GRD5-like element there was amplification and divergence of GRD5 units before the ancestor diverged into different species. The GRD3 family appears to have arisen later in section Tomentosae speciation in a lineage of N. tomentosiformis (Murad et al, 2002). Once again there was amplification and divergence of GRD3 units that occurred before the formation of tobacco (Figure 5). Alternatively, and less likely, there could have been horizontal transfer of GRD sequences between taxa resulting in the same pattern.

GRD5 integrated into the Nicotiana genome after the divergence of N. otophora (a), presumably by illegitimate recombination with a free-living geminivirus (Ashby et al, 1997). The sequence became methylated, amplified and diversified into the GRD5 family before there was further Tomentosae speciation. After divergence within N. tomentosiformis (b), another family evolved, probably via a second independent integration event involving another geminivirus, an endogenous Helitron and GRD5, to form the GRD3 family at a second chromosomal locus. This line of N. tomentosiformis went on to form tobacco (c), on hybridisation and allopolyploidy involving N. sylvestris.
The clone GRD53 was considered in earlier works to be distinctive (Bejarano et al, 1996), and uniquely the clone labelled both the GRD3 sequences (strongly) and the GRD5 sequences (weakly) by Southern and in situ hybridisation. However, on the phylogenetic reconstruction, the sequence clearly represents a divergent member of the GRD3 family of sequences and indeed was originally cloned in tandem array with GRD3 (Bejarano et al, 1996).
GRD integration is polyphyletic
A comparison of synonymous vs nonsynonymous nucleotide substitutions across all classes of GRD reveals a significant indication of purifying selection acting at the level of the gene product. Perhaps translation of a truncated dominant-negative viral rep sequence initially gave some resistance to virus infection. Another possible mode of selection could have acted if GRDs were involved in post-transcriptional gene silencing (Baulcombe, 2001), so elevating the host plant's resistance to infection through RNA:RNA duplex formation with viral rep RNA, that is, exactly the effect that Lichtenstein and colleagues were engineering when GRD was discovered (Bejarano et al, 1996). But this type of selection does not easily explain substitution bias because it acts at the nucleotide sequence level rather than at the amino-acid sequence. And if such a mechanism occurred at all, it is likely that the high rate of viral divergence would have rapidly eroded any selective advantage conferred by GRD.
When elements within each GRD family are compared separately, the rates are close to even, and show no significant sign of purifying selection. One possible explanation is that selection was lost at/after the point divergence of the two families. Since the GRD3 family integrated later than GRD5 in the evolution of Nicotiana, and after GRD5 divergence, perhaps a copy of GRD5 transposed from chromosome 4 to chromosome 2 in a lineage of N. tomentosiformis. This scenario predicts that the GRD3 family would be most similar to a particular GRD5 element, but no such ancestral element has been found and indeed the GRD3 family carries additional sequences (see below) not found in the GRD5 family.
One explanation that does fully reconcile the available data is that there were in fact two integration events, separated by time and involving geminiviruses that are either extinct or not represented on the phylogenetic scheme. The data suggest that the first integration gave rise to the GRD5 family. The second integration event, occurring after the divergence of Tomentosae on a different chromosome, gave rise to the GRD3 family (Figure 5). This could have involved some form of recombination between GRD5, geminiviral rep and perhaps a mobile Helitron (see Figure 4). During the period between the two integration events, when the progenitor geminivirus was ‘free living’, most of its nonsynonymous mutations would have been selected against in rep (Figure 6). Integration via recombination with pre-existing GRD is plausible because recombination between viruses and genomic DNA is established for retroviruses, and may also occur within DNA viruses. The fact that the geminivirus is unknown to us is unsurprising since most geminiviruses examined are agricultural geminiviruses with a serious pathology. Geminiviruses of wild plants have simply not been the subject of much interest.

A model for double integration of GRD that is consistent with both the distribution of GRD in extant species of Nicotiana and the signature of purifying selection in the ancestors of modern GRD sequences. Parameter ω is the ratio of nonsynonymous to synonymous substitution rates, and is a precise measure of election pressure. Values of ω<1 indicate purifying selection, whereas ω=1 indicates neutral evolution.
References
Ashby MK, Warry A, Bejarano ER, Khashoggi A, Burrell M, Lichtenstein CP (1997). Analysis of multiple copies of geminiviral DNA in the genome of four closely related Nicotiana species suggest a unique integration event. Plant Mol Biol 35: 313–321.
Baulcombe D (2001). RNA silencing – diced defence. Nature 409: 295–296.
Bejarano ER, Khashoggi A, Witty M, Lichtenstein C (1996). Integration of multiple repeats of geminiviral DNA into the nuclear genome of tobacco during evolution. Proc Natl Acad Sci USA 93: 759–764.
Bejarano ER, Lichtenstein CP (1994). Expression of TGMV antisense RNA in transgenic tobacco inhibits replication of BCTV but not ACMV geminiviruses. Plant Mol Biol 24: 241–248.
Bielawski JP, Dunn KA, Yang ZH (2000). Rates of nucleotide substitution and mammalian nuclear gene evolution: approximate and maximum-likelihood methods lead to different conclusions. Genetics 156: 1299–1308.
Bird AP (1986). CpG-rich islands and the function of DNA methylation. Nature 321: 209–213.
Brough CL, Gardiner WE, Inamdar NM, Zhang XY, Ehrlich M, Bisaro DM (1992). DNA methylation inhibits propagation of tomato golden mosaic-virus DNA in transfected protoplasts. Plant Mol Biol 18: 703–712.
Day AG, Bejarano ER, Buck KW, Burrell M, Lichtenstein CP (1991). Expression of an antisense viral gene in transgenic tobacco confers resistance to the DNA virus tomato golden mosaic-virus. Proc Natl Acad Sci USA 88: 6721–6725.
Dunn KA, Bielawski JP, Yang ZH (2001). Substitution rates in Drosophila nuclear genes: implications for translational selection. Genetics 157: 295–305.
Felsenstein J (1985). Confidence-limits on phylogenies – an approach using the bootstrap. Evolution 39: 783–791.
Feschotte C, Wessler SR (2001). Treasures in the attic: rolling circle transposons discovered in eukaryotic genomes. Proc Natl Acad Sci USA 98: 8923–8924.
Frederico LA, Kunkel TA, Shaw BR (1990). A sensitive genetic assay for the detection of cytosine deamination – determination of rate constants and the activation-energy. Biochemistry 29: 2532–2537.
Gibbs MJ, Weiller GF (1999). Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc Natl Acad Sci USA 96: 8022–8027.
Goldman N, Yang ZH (1994). Codon-based model of nucleotide substitution for protein-coding DNA-sequences. Mol Biol Evol 11: 725–736.
Hanley-Bowdoin L, Settlage SB, Orozco BM, Nagar S, Robertson D (2000). Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation. Crit Rev Biochem Mol Biol 35: 105–140.
Hasegawa M, Kishino H, Yano TA (1985). Dating of the human ape splitting by a molecular clock of mitochondrial-DNA. J Mol Evol 22: 160–174.
Higgins DG, Sharp PM (1998). CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 73: 237–244.
Kapitonov VV, Jurka J (2001). Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA 98: 8714–8719.
Kimura M (1983). The Neutral Theory of Evolution. Cambridge University Press: Cambridge, UK.
Koonin EV, Ilyina TV (1992). Geminivirus replication proteins are related to prokaryotic plasmid rolling circle DNA-replication initiator proteins. J Gen Virol 73: 2763–2766.
Kovarik A, Koukalova B, Lim KY, Matyasek R, Lichtenstein CP, Leitch AR et al (2000). Comparative analysis of DNA methylation in tobacco heterochromatic sequences. Chromosome Res 8: 527–541.
Kovarik A, Matyasek R, Leitch A, Gazdova B, Fulnecek J, Bezdek M (1997). Variability in CpNpG methylation in higher plant genomes. Gene 204: 25–33.
Lim KY, Matyasek R, Lichtenstein CP, Leitch AR (2000). Molecular cytogenetic analyses and phylogenetic studies in the Nicotiana section Tomentosae. Chromosoma 109: 245–258.
Murad L, Lim KY, Christopodulou V, Matyasek R, Lichtenstein CP, Kovarik A (2002). The origin of tobacco's T genome is traced to a particular lineage within Nicotiana tomentosiformis (Solanaceae). Am J Bot 89: 921–928.
Penny D, Hendy MD (1985). Testing methods of evolutionary tree construction. Cladistics 1: 266–272.
Swofford DL (1993). PAUP: Phylogenetic Analysis using Parsimony. version 3.1.1 Computer Programme. Illinois Natural History Survey: Champaign, IL.
Yang ZH (1994). Statistical properties of the maximum-likelihood method of phylogenetic estimation and comparison with distance matrix-methods. Syst Biol 43: 329–342.
Yang ZH (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences 13: 555–556.
Yang Z, Bielawski JP (2000). Statistical methods for detecting molecular adaptation. Trends in Ecology and Evolution 15: 496–503.
Yang ZH, Nielsen R (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17: 32–43.
Yoder JA, Walsh CP, Bestor TH (1997). Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335–340.
Zingg JM, Shen JC, Jones PA (1998). Enzyme-mediated cytosine deamination by the bacterial methyltransferase M.MspI. Biochem J 332: 223–230.
Acknowledgements
We thank NERC, The Royal Society and The Grant Agency of the Czech Republic (No. 521/98/0045) for support of this work. JPB was supported by a Biotechnology and Biological Sciences Research Council Grant (31/G10434). We thank Queen Mary for a College studentship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murad, L., Bielawski, J., Matyasek, R. et al. The origin and evolution of geminivirus-related DNA sequences in Nicotiana. Heredity 92, 352–358 (2004). https://doi.org/10.1038/sj.hdy.6800431
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800431
Keywords
This article is cited by
-
Virus-mediated export of chromosomal DNA in plants
Nature Communications (2018)
-
Exploring the genetic diversity and molecular evolution of fig badnavirus-1 from Iran
Journal of Plant Pathology (2018)
-
Reliability of the nanopheres-DNA immunization technology to produce polyclonal antibodies directed against human neogenic proteins
Molecular Genetics and Genomics (2013)
-
Widespread Horizontal Gene Transfer from Circular Single-stranded DNA Viruses to Eukaryotic Genomes
BMC Evolutionary Biology (2011)
-
Interkingdom protein domain fusion: the case of an antimicrobial protein in potato (Solanum tuberosum)
Plant Systematics and Evolution (2011)
