
Abstract
Background:
Pralatrexate is a dihydrofolate reductase (DHFR) inhibitor with high affinity for reduced folate carrier 1 (RFC-1) and folylpolyglutamate synthetase (FPGS), resulting in extensive internalization and accumulation in tumour cells. Pralatrexate is approved in the US for the treatment of relapsed or refractory peripheral T-cell lymphoma and is being investigated in various malignancies. Here, we evaluated molecular correlates of sensitivity to pralatrexate and explored combinations with a variety of anticancer agents.
Methods:
Antiproliferative effects of pralatrexate were evaluated in 15 human-cancer cell lines using the MTT assay. Gene expression was evaluated using qRT–PCR.
Results:
Pralatrexate and methotrexate had a similar pattern of cytotoxicity, pralatrexate being more potent. Pralatrexate potentiated the effects of platinum drugs, antimetabolites and EGFR inhibitors. Dose- and time-dependent cytotoxicity of pralatrexate correlated with high mRNA expression of FPGS. Acquired resistance to pralatrexate was associated with decreased RFC-1 expression, whereas methotrexate resistance correlated with increased DHFR expression, suggesting different mechanisms of acquired resistance.
Conclusion:
Pralatrexate was more potent than methotrexate in a panel of solid tumour lines. Our findings support the further clinical development of pralatrexate in combination with certain cytotoxics and targeted therapies, and suggest that RFC-1, FPGS and DHFR may be potential biomarkers of outcome.
Similar content being viewed by others

Main
The folate pathway has a key role in cell growth and proliferation (Appling, 1991; Odin et al, 2003). Folic acid (folate) enters cells through reduced folate carrier 1 (RFC-1), is polyglutamated by folylpolyglutamate synthetase (FPGS), and is reduced to dihydrofolate, which is further converted to tetrahydrofolate (THF) by dihydrofolate reductase (DHFR). The different enzymes and transporters involved in this pathway are targets for an important class of cytotoxic agents, antifolates. Methotrexate was one of the first agents of this class and was first used in the treatment of childhood acute lymphoblastic leukaemia (Farber et al, 1948). Since then, methotrexate has been widely used in haematologic and solid cancers, and new generations of antifolates have been rationally designed to exploit multiple aspects of the folate pathway (e.g., raltitrexed in colorectal cancer (Cocconi et al, 1998), and pemetrexed in malignant pleural mesotheliomas (Vogelzang et al, 2003) and non-small-cell lung carcinomas (NSCLC) (Hanna et al, 2004)).
Pralatrexate ((RS)-10-propargyl-10-deazaaminopterin) is a synthetic anti-DHFR antifolate, rationally designed to have greater affinity for RFC-1 and FPGS, resulting in increased cytotoxic activity as compared with methotrexate (DeGraw et al, 1993; Sirotnak et al, 1998). The cytotoxicity of pralatrexate was shown to be correlated with RFC-1 mRNA expression in human lymphoma cells (Wang et al, 2003).
Pralatrexate is undergoing clinical evaluation as a single agent or in combination in several tumour types, including lymphoma (O’Connor et al, 2007; Marneros et al, 2009), and has received accelerated approval by the US Food and Drug Administration (FDA) for patients with refractory or relapsed peripheral T-cell lymphoma.
In this study, we have compared the antiproliferative activity of pralatrexate with that of methotrexate and other antimetabolites in several human solid tumour cell lines. In addition, we further evaluated molecular mechanisms of action of pralatrexate, and screened for markers of sensitivity and acquired resistance to pralatrexate, knowledge of which could be of potential use for designing future clinical trials. Finally, possible sequence-dependent synergy or additive effects of pralatrexate with various cytotoxic and targeted agents were also investigated.
Materials and methods
Cell lines
A panel of colon (HT29, HCT116, COLO205 and HCC2998), breast (MCF7), melanoma (MDA-MB-435, formerly designated as a breast cancer line), NSCLC (HOP62 (adenocarcinoma) and HOP92 (large cell carcinoma)), ovarian (OVCAR3, IGROV1), prostate (DU145, PC3) and head and neck (SCC61, HEP2 and SQ20B) cancer cell lines was purchased from the ATCC (Rockville, MD, USA) and National Cancer Institute collections. Cells were grown as monolayers in RPMI medium supplemented with 10% fetal calf serum (Invitrogen, Cergy-Pontoise, France), 2 mM glutamine, 100 U ml−1 penicillin and 100 μ M ml−1 streptomycin. In this study, we used unpurified media potentially containing glycine, hypoxanthine and thymidine, which may have theoretically reduced drug activity. However, considering the high levels of resistance developed in our cell lines and taking into account that both parental and derived resistant counterpart were grown in similar media, it remains unlikely that this would have severely impact on data.
Cytotoxicity assays
Cell viability was determined using the MTT assay, which was carried out as described previously (Hansen et al, 1989). Briefly, cells were seeded in 96-well plates at a density of 2 × 103 cells per well. Cells were incubated for 120 h and then 0.4 mg ml−1 of MTT dye (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; Sigma, Saint-Quentin Fallavier, France), was added for 4 h at 37°C. Media was removed and the monolayer suspended in 0.1 ml of DMSO, after which the absorbance at 560 nm was measured using a microplate reader (Thermo, Saint-Herblain, France). The control value corresponding to untreated cells was defined as 100% and the viability of treated samples was expressed as a percentage of the control. IC50 values were determined as concentrations that reduced cell viability by 50%.
For single agent studies, cells were treated with increasing concentrations of pralatrexate, methotrexate, pemetrexed, 5-FU and 5′-DFUR for up to 72 h. Then, the cells were allowed to recover in compound-free medium for 48 h before determination of growth inhibition using the MTT assay.
For combination studies of pralatrexate with other anticancer drugs, sequential (pralatrexate for 24 h, followed by the second agent for 24 h or the inverse schedule) or simultaneous (exposure to both agents for 24 h) schedules were used. Cells were treated with various concentrations of the drugs and the combinations. Growth inhibition was then determined by the MTT assay.
Statistical analysis and determination of synergistic activity
Drug combination effects were determined using the Chou and Talalay method (Chou and Talalay, 1984) based on the median effect principle. Combination index (CI) values of <1 indicate synergy, a CI value of 1 indicates additive effects and a CI value of >1 indicates antagonism. Data were analysed using the concentration-effect analysis software (Biosoft, Cambridge, UK). For statistical analysis and graphs, the Instat and Prism software (GraphPad, San Diego, CA, USA) were used. Experiments were performed three times, in duplicate. Means and standard deviations were compared using the Student's t-test (two-sided P-value).
Cell cycle analysis and apoptosis
The cell cycle stage and percentage of apoptotic cells were assessed by flow cytometry. In brief, cells were treated with various concentrations of pralatrexate, fixed in 70% ethanol and stored at −20°C until use. Cells were rehydrated in PBS, incubated for 30 min at 37°C with 250 μg ml−1 RNAse A and 20 min at 4°C with 50 μg ml−1 propidium iodide in the dark. The cell cycle distribution and percentage of apoptotic cells were determined with a FACScan flow cytometer and analyzed by FACS Calibur (Becton Dickinson, Le Pont de Claix, France). Apoptosis induction was evaluated using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Le Pont de Claix, France).
Gene expression analysis by RT–PCR
The theoretical and practical aspects of quantitative real-time PCR (qRT–PCR) using the ABI Prism 7900 Sequence Detection System (Perkin-Elmer Applied Biosystems, Foster City, CA, USA) have been described in detail elsewhere (Bièche et al, 2001). Results were expressed as fold differences in target gene expression relative to the TBP gene (an endogenous RNA control) and relative to a calibrator (1 × sample), consisting of the cell line sample from our tested series that contained the smallest amount of target gene mRNA. Experiments were performed in duplicate.
Western blot analysis
Cells were lysed in buffer containing 50 mM HEPES (pH 7.6), 150 mM NaCl, 1% Triton X-100, 2 mM sodium vanadate, 100 mM NaF and 0.4 mg ml−1 phenylmethylsulfonyl fluoride. Equal amounts of protein (20–50 μg per lane) were subjected to SDS–PAGE and transferred to nitrocellulose membranes. Membranes were probed with anti-cleaved PARP, anti-cleaved caspase 3, anti-caspase 9 (Cell Signaling, Saint Quentin Yvelines, France), anti-DHFR (Abcam, Paris, France), anti-β-actin (Sigma-Aldrich, Saint-Quentin Fallavier, France)-specific primary antibodies, followed by peroxidase-linked secondary antibodies and visualisation by chemiluminescence.
Results
Single-agent antiproliferative effects
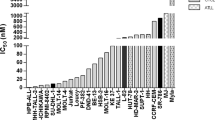
The antiproliferative effects of pralatrexate were examined in 15 cancer cell lines as displayed in Table 1. Time course experiments showed that optimal antiproliferative effects were achieved when cells were exposed to pralatrexate for 72 h (Figure 1A). Pralatrexate IC50 values ranged from 0.01±0.002 μ M for the prostate cancer cell line PC3 to >350 μ M for the MDA-MB-435 cell line. Interestingly, two groups of cell lines with more than 100-fold difference in IC50 were observed: One group including PC3, SCC61, DU145, HT29, HOP62, SQ20B, HOP92, HEP2 and IGROV1 cells displayed IC50 <0.1 μ M, whereas Colo205, HCC2998, MCF7, HCT116, OVCAR3 and MDA-MB-435 cells showed IC50 values ⩾9 μ M.

Pralatrexate cytotoxicity in a panel of human cancer cell lines. (A) Pralatrexate (PDX) time-course (IC50 values, 1, 5, 24 and 72 h drug exposure) cytotoxicity in sensitive cell lines. (B) Comparative analysis of 72 h cytotoxicity of pralatrexate, methotrexate, pemetrexed, 5-FU and 5′-DFUR in a panel of cancer cell lines. The indicated values are calculated as follows: log (IC50 individual cell line) – mean (log IC50). Negative values indicate that the cell line is more sensitive than the average, whereas positive values indicate that the cell line is more resistant than the average.
The antiproliferative effects of pralatrexate were compared with those of methotrexate and several commonly used antimetabolites such as pemetrexed, 5-FU and 5′-DFUR, the active capecitabine metabolite (Figure 1B and Table 1). Pralatrexate IC50 values were on average almost 10-fold lower than those observed for methotrexate. The cytotoxicity profiles of these two antifolates were similar with the same distinct groups of sensitive and resistant cell lines. The cytotoxicity profile of pralatrexate was different from that of 5-FU, 5′-DFUR and pemetrexed, suggesting differences in the metabolism, mechanism of action and/or resistance between pralatrexate and other antimetabolites. Interestingly, limited cross-sensitivity was observed between pralatrexate and pemetrexed, an antifolate, believed to be primarily a thymidylate synthetase (TS) inhibitor.
Effect of pralatrexate and methotrexate on cell cycle changes and apoptosis
The prostate cancer cell line DU 145 was selected as a sensitive model to both pralatrexate and methotrexate for further investigation in this study. Cells were incubated with concentrations of 0.1 μ M (IC50) and 0.2 μ M (two-fold IC50) of pralatrexate and 0.6 μ M (IC50) and 1.2 μ M (two-fold IC50) of methotrexate for 24 h. Cell cycle analysis showed that pralatrexate-treated cells had decreased proportions of cells in S and G2/M phases with an increase of sub-G1 fraction (>eight-fold), suggesting apoptosis induction (Figure 2A). A similar pattern, albeit less pronounced, was observed in methotrexate-treated cells (Figure 2A). Thus, both agents caused the accumulation in the G0/G1 phase and possible apoptosis induction.

Effects of pralatrexate (PDX) and methotrexate (MTX) (24 h exposure) on cell cycle distribution (A), apoptosis induction (Annexin V staining) (B) and activation of PARP, caspase 3 and caspase 9 (C) in DU145 prostate cancer cells. *Significant difference (P<0.05) comparing with control.
Annexin V/PI double staining showed that apoptosis increased 2.6- and 4-fold in DU145 cells at concentrations of 0.1 and 0.2 μ M pralatrexate, respectively. Similar results, with 1.5- and 2-fold increase in apoptosis, were observed in DU145 cells treated with 0.6 and 1.2 μ M methotrexate, respectively, again showing superior activity of pralatrexate compared with methotrexate. Apoptosis induction by pralatrexate was further confirmed by increases in cleaved PARP and caspases observed after 24 h exposure to pralatrexate (see Figure 2C). From these experiments it appears that apoptosis is the major mechanism of cancer cell death induced by pralatrexate in this model.
Expression of genes involved in folate transport and metabolism
The expression of genes known to be involved in sensitivity to antifolates was analyzed in the panel of cancer cell lines. DHFR, FPGS, TS/TYMS, thymidylate synthetase, SCL19A1/RFC-1, GARFT (glycinamide ribonucleotide formyl transferase), SLC25A32 (mitochondrial folate transporter/carrier) and ABC transporter B1 (ABCB1 or MDR1) mRNA expression was determined by qRT–PCR (Figure 3A). The cell lines expressed various levels of these folate pathway genes but no significant correlation was found between sensitivity to pralatrexate and mRNA expression of TS, SCL19A1/RFC-1, GARFT, SLC25A32 and MDR1. Pralatrexate-sensitive cells expressed relatively higher levels of DHFR, a target of pralatrexate, than the ‘resistant’ group, but this did not reach statistical significance (P=0.083, Figure 3A). Pralatrexate-sensitive cells expressed significantly higher levels of FPGS mRNA than resistant cells (t-test, P=0.002). Overall, a trend towards a positive correlation between FPGS mRNA expression and pralatrexate sensitivity (IC50 values) was found (R2=0.47, P<0.01), suggesting an important role of polyglutamation in pralatrexate antiproliferative activity.

Relative mRNA expression of folate genes in pralatrexate sensitive and -resistant cell lines. (A) Relative expression of DHFR, FPGS, TS, GART, SLC25A32, SCL19A1/RFC-1 and ABCB1/MDR1 mRNA in sensitive- and resistant groups. (B) Correlation between pralatrexate sensitivity (IC50 values) and mRNA expression of SCL19A1/RFC-1 and SLC25A32 folate transporters in nine pralatrexate-sensitive cell lines.
To determine the potential role of folate transporters in pralatrexate activity, we correlated the IC50 values obtained after 72 h drug exposure with the level of mRNA expression of SCL19A1/RFC-1 and SLC25A32 in the nine pralatrexate-sensitive cell lines (Figure 3B). Cells that expressed a high level of SCL19A1/RFC-1 and SLC25A32 mRNA displayed higher sensitivity to pralatrexate, suggesting potential roles of SCL19A1/RFC-1 and SLC25A32 in cellular uptake of pralatrexate.
Development of pralatrexate and methotrexate resistant cell lines
To characterize potential mechanisms of resistance to pralatrexate, the cell lines DU-PDX and HEP-PDX were developed from parental DU145 and HEP2 cells, respectively, by exposure to increasing concentrations of pralatrexate over a period of 6 months. DU-PDX and HEP-PDX cells were at least 200- and 500-fold less sensitive to pralatrexate than parental cells (Figure 4A). After five passages in drug-free medium, the resistant cells retained their drug resistance, suggesting stability of these cell lines. As shown in Figure 4A, pralatrexate-resistant cell lines showed partial cross-resistance to methotrexate.

Characterization of pralatrexate-resistant cell lines. (A) Pralatrexate (top panel) and methotrexate (bottom panel) cytotoxicity in DU-PDX and HEP-PDX cell lines compared with their parental counterparts DU145 and HEP2. (B) Relative mRNA expression of folate genes in pralatrexate-resistant cell lines. (C) Western blot of DHFR protein in DU145 and HEP2 sensitive and DU-PDX, DU-MTX, HEP-PDX and HEP-MTX pralatrexate and methotrexate-resistant cell lines.
To compare the mechanisms of pralatrexate and methotrexate resistance, methotrexate-resistant cell lines DU-MTX and HEP-MTX were developed. DU-MTX and HEP-MTX displayed cross-resistance to pralatrexate, however, the activity of pralatrexate still remained superior (approximately 10-fold lower IC50) to that of methotrexate (Data not shown).
Genetic changes associated with acquired pralatrexate resistance
To determine possible mechanisms of anti-folate resistance, we evaluated the mRNA expression of several folate genes in parental and resistant cells. As shown in Figure 4B, mRNA expression of DHFR, TS and SLC25A32 was not significantly changed in pralatrexate-resistant cells. A slight decrease in FPGS mRNA expression was observed in DU-PDX and HEP-PDX cells compared with their parental counterparts. In contrast, SCL19A1/RFC-1 mRNA expression was >10-fold decreased in the two pralatrexate-resistant cell lines. mRNA level of ABCB1/MDR1 was 40- and 2-fold higher in DU-PDX and HEP-PDX, respectively, compared with DU145 and HEP2. These data suggest an important role of transporters in pralatrexate antiproliferative activity and acquired resistance. To study the role of MDR1 in pralatrexate resistance, DU-PDX and HEP-PDX cells were incubated with 30 μ M verapamil, a competitive substrate of MDR1, and 3 μ M cyclosporin A concomitantly with pralatrexate for 72 h. No changes were observed in pralatrexate cytotoxicity with and without verapamil and cyclosporine A, suggesting that MDR1 overexpression may not have a major role in acquired resistance to pralatrexate in these cell lines.
Analysis of expression of DHFR, a target of pralatrexate and methotrexate, showed significant increases in mRNA (more that 30-fold), as well as >10-fold increases in DNA gene copy numbers. DHFR protein expression in HEP-MTX cells was consistently higher compared with parental HEP2 cells (Figure 4C), suggesting that DHFR amplification has an important role in resistance to methotrexate (Figure 4C). Slight increase of DHFR expression in DU-PDX and HEP-PDX was nonsignificant as compared with that in parental DU145 and HEP2 cells (P=0.083). This suggested that the molecular mechanism of acquired resistance to pralatrexate in HEP-PDX cells might differ from acquired methotrexate resistance in HEP-MTX cells.
Analysis of cross-resistance to other antifolates and antimetabolites
To evaluate the cross-resistance of pralatrexate-resistant cells to other drugs, DU145, DU-PDX, HEP2 and HEP-PDX cells were exposed to pemetrexed and 5-FU for 72 h. No significant difference between parental and PDX-resistant cells was observed for 5-FU cytotoxicity. Pemetrexed exposure for 72 h was only slightly less cytotoxic in DU-PDX and HEP-PDX cells compared with their parental counterparts (data not shown). These data suggest that acquired resistance to pralatrexate may not translate into resistance to pemetrexed and 5-FU, possibly because of the differences in mechanism of action of these compounds.
Combination of pralatrexate with anticancer drugs
The effects of sequential and simultaneous exposure of combinations of pralatrexate with oxaliplatin, cisplatin, 5-FU, 5′-DFUR, SN38, paclitaxel, gemcitabine, erlotinib or lapatinib were evaluated in DU145 cells (Table 2).
The combination of pralatrexate with oxaliplatin or cisplatin resulted in synergistic effects (CI<1) when drugs were given simultaneously irrespective of the concentrations used. Pralatrexate given before oxaliplatin demonstrated only additive effects and was synergistic with cisplatin for the same administration schedule. Administration of either of the platinum drugs before pralatrexate was not beneficial. Thus, the effects of pralatrexate in combination with platinum drugs appear to be schedule dependent.
The combination of pralatrexate with the antimetabolites 5-FU or 5′-DFUR resulted in synergistic effects for the sequential administration schedules and appears to be antagonistic when the drugs were administered simultaneously. Combination of pralatrexate with SN38 (the active metabolite of irinotecan) in DU145 cells resulted in synergistic effects for all schedules of administration. The combination of pralatrexate with paclitaxel resulted in some synergistic effects for the sequential administration schedule when paclitaxel was given before pralatrexate and additive/antagonistic for the reverse sequence and simultaneous exposure. The combinations of pralatrexate with erlotinib or lapatinib were shown to be synergistic when pralatrexate was given first or simultaneously with these EGFR kinase inhibitors, suggesting a role of EGFR signalling in response to pralatrexate.
Discussion
Pralatrexate is an antifolate with high affinity for the reduced folate carrier 1 (RFC-1) protein and folylpolyglutamate synthetase (FPGS), resulting in extensive internalization and accumulation within tumour cells. Pralatrexate is currently being investigated as a single agent and in combinations in various malignancies. In order to guide further clinical development, molecular correlates of sensitivity to pralatrexate and preclinical data on combination treatments are needed.
Pralatrexate displayed potent antiproliferative activity (IC50 <0.1 μ M) in 9 out of the 15 human solid-tumour cell lines. Two distinct groups of cell lines were identified with >100-fold difference in pralatrexate IC50 values: sensitive and relatively resistant cell lines. The in vitro antiproliferative effects of pralatrexate in terms of IC50 values were on average almost 10-fold better than those observed with methotrexate. When comparing the cytotoxic activity of these two similar antifolates to other antimetabolites including 5-FU, 5′-DFUR and pemetrexed, pralatrexate appears to retain activity in several cells that were poorly sensitive to 5-FU and 5′-DFUR, such as NSCLC HOP62 and HOP92 cell lines. Similarly, the sensitivity profile for pemetrexed was different from that for pralatrexate, which may be explained by the differences in molecular mechanism of action of these compounds.
Izbicka et al (2009) have shown that pralatrexate demonstrated unique attributes relative to methotrexate and pemetrexed. Pralatrexate exhibited enhanced cellular uptake and increased polyglutamation, which correlated with increased tumour-growth inhibition in a NSCLC xenograft model. A positive correlation between pralatrexate sensitivity and mRNA expression of FPGS, a major enzyme responsible for polyglutamation of antifolates, was found in the 15 cancer cell lines, suggesting an important role of polyglutamation in cellular response to pralatrexate. It was shown (Chabner et al, 1985) that if drug exposure is limited to brief periods (24 h or less), cell lines that do not form polyglutamates efficiently are insensitive to antifolate drugs. These short exposure conditions approximate clinical chemotherapy exposure and emphasize the importance of rapid production of the polyglutamated metabolites for cytotoxicity. Continuous exposure to drug may diminish the importance of polyglutamation. In our study, short (1 and 5 h) pralatrexate exposure induced pronounced antiproliferative effects and these effects may be due to improved pralatrexate cellular uptake and polyglutamation by FPGS. Cellular uptake is likely dependent on expression of folate carriers such as RFC-1 and SLC25A32. Although a positive correlation between pralatrexate sensitivity and mRNA expression of FPGS, was found in the panel of 15 cancer cell lines, only a slight but nonsignificant decrease in FPGS mRNA expression was observed in DU-PDX and HEP-PDX cells with acquired resistance to pralatrexate compared with their parental counterparts suggesting that polyglutamation may not be regarded as a main predictive factor of pralatrexate activity .
To better elucidate the predictive genetic factors of pralatrexate sensitivity, two cell lines with acquired resistance to the drug were developed from DU145 (prostate) and HEP2 (head and neck) cancer cell lines. Being more than 200-fold more resistant to pralatrexate than parental cells, DU-PDX and HEP-PDX displayed partial cross-resistance to methotrexate. Pralatrexate acquired resistance was associated with decreased RFC-1 expression and increased MDR1 expression. Fotoohi et al (2009) described antifolate-resistant leukaemia lines with mRNA levels of RFC-1 downregulated more than two-fold in methotrexate-resistant cells, emphasizing the important role of inux transport in antifolate resistance. Similar data were previously obtained by Jansen and Pieters (1998), Rothem et al (2004), and Ifergan et al (2003) using other methotrexate-resistant cellular models. Increased MDR1 expression does not appear to have a role in the observed acquired resistance to pralatrexate as inhibition of MDR1 did not restore sensitivity to pralatrexate. Decreased FPGS activity was shown to be associated with acquired resistance to methotrexate in human leukaemia CCRF-CEM cells (Mauritz et al, 2002). In our study, a slight decrease in FPGS expression was observed in pralatrexate-resistant cells, suggestive of a role for polyglutamation in resistance to pralatrexate. Studies performed three decades ago discovered that another frequent mechanism of acquired methotrexate resistance is DHFR gene amplification and the consequent enzyme overexpression (reviewed by Chen et al, 1995; Assaraf, 2007). Indeed, in our study, the cell line HEP-MTX, with acquired resistance to methotrexate, displayed a dramatic increase in DHFR mRNA and protein expression as compared with its parental counterpart. Increased DHFR expression was not observed in the pralatrexate-resistant cell lines. Although differences were not statistically significant, pralatrexate-sensitive cells expressed relatively higher levels of DHFR, a putative target of pralatrexate, than resistant cells. These data suggest that expression of DHFR may not be a major factor in pralatrexate sensitivity. These findings suggest different molecular mechanisms of resistance to methotrexate and pralatrexate in these cell lines.
The value of antifolates as anticancer treatments is potentiated by their use in drug combinations. A study of pralatrexate administration in combination with gemcitabine in a panel of lymphoma cell lines demonstrated not only that this combination is synergistic and more efficient than methotrexate/gemcitabine in generating apoptosis but also that the effects were highly sequence dependent (Toner et al, 2006). Pralatrexate has also been administered in combination with taxanes in a phase-I clinical trial, resulting in significant antitumour activity (Azzoli et al, 2007). To provide further insight into the potential clinical use of pralatrexate in patients with cancer, we combined pralatrexate with several classical anticancer drugs including cisplatin, oxaliplatin, 5-FU, 5′-DFUR, gemcitabine, paclitaxel, SN38 and the EGFR tyrosine kinase inhibitors erlotinib and lapatinib. Pralatrexate demonstrated synergistic effects with several anticancer agents and these effects were schedule dependent. Highly synergistic effects were observed by Toner, when pralatrexate was administered before gemcitabine in lymphoma models (Toner et al, 2006). In our experiments, simultaneous exposure to pralatrexate with gemcitabine was one of the best schedules. However, these results may be model dependent. Interestingly, our results also displayed synergistic antiproliferative effects when pralatrexate was given before or simultaneously with the EGFR inhibitor erlotinib, or the dual EGFR/HER2 inhibitor lapatinib.
In summary, pralatrexate was shown to be more potent than other antifolates in a panel of solid tumour models. Several genetic factors including RFC-1, FPGS and DHFR may be regarded as predictive factors of pralatrexate activity in solid tumour cells. Pralatrexate showed synergistic cytotoxic activity with several classical cytotoxic agents as well as with targeted EGFR inhibitors in the DU-145 prostate cancer line, and further investigation in clinical trials is warranted.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Appling DR (1991) Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J 5: 2645–2651
Assaraf YG (2007) Molecular basis of antifolate resistance. Cancer Metastasis Rev 26: 153–181
Azzoli CG, Krug LM, Gomez J, Miller VA, Kris MG, Ginsberg MS, Henry R, Jones J, Tyson L, Dunne M, Pizzo B, Farmer A, Venkatraman E, Steffen R, Sirotnak FM (2007) A phase 1 study of pralatrexate in combination with paclitaxel or docetaxel in patients with advanced solid tumors. Clin Cancer Res 13: 2692–2698
Bièche I, Parfait B, Laurendeau I, Girault I, Vidaud M, Lidereau R (2001) Quantification of estrogen receptor alpha and beta expression in sporadic breast cancer. Oncogene 20: 8109–8115
Chabner BA, Allegra CJ, Curt GA, Clendeninn NJ, Baram J, Koizumi S, Drake JC, Jolivet J (1985) Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest 76: 907–912
Chen G, Wright JE, Rosowsky A (1995) Dihydrofolate reductase binding and cellular uptake of nonpolyglutamatable antifolates: correlates of cytotoxicity toward methotrexate-sensitive and -resistant human head and neck squamous carcinoma cells. Mol Pharmacol 48: 758–765
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55
Cocconi G, Cunningham D, Van Cutsem E, Francois E, Gustavsson B, van Hazel G, Kerr D, Possinger K, Hietschold SM (1998) Open, randomized, multicenter trial of raltitrexed vs fluorouracil plus high-dose leucovorin in patients with advanced colorectal cancer. Tomudex Colorectal Study Group. J Clin Oncol 16: 2943–2952
DeGraw JI, Colwell WT, Piper JR, Sirotnak FM (1993) Synthesis and antitumor activity of 10-propargyl-10-deazaaminopterin. J Med Chem 36: 2228–2231
Farber S, Diamond LK, Mercer RD, Sylvester RF, Wolff JA (1948) Temporary remission in acute leukaemia in children produced by folio acid antagonist 4-aminopteroylglutamic acid (aminopterin). New England J Med 238: 787–793
Fotoohi AK, Assaraf YG, Moshfegh A, Hashemi J, Jansen G, Peters GJ, Larsson C, Albertioni F (2009) Gene expression profiling of leukemia T-cells resistant to methotrexate and 7-hydroxymethotrexate reveals alterations that preserve intracellular levels of folate and nucleotide biosynthesis. Biochem Pharmacol 77: 1410–1417
Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, Lim HL, Desch C, Szondy K, Gervais R, Shaharyar M, Manegold C, Paul S, Paoletti P, Einhorn L, Bunn Jr PA (2004) Randomized phase III trial of pemetrexed vs docetaxel in patients with non-small cell lung cancer previously treated with chemotherapy. J Clin Oncol 22: 1589–1597
Hansen MB, Nielsen SE, Berg K (1989) Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 119: 203–210
Ifergan I, Meller I, Issakov J, Assaraf YG (2003) Reduced folate carrier protein expression in osteosarcoma: implications for the prediction of tumor chemosensitivity. Cancer 98: 1958–1966
Izbicka E, Diaz A, Streeper R, Wick M, Campos D, Steffen R, Saunders M (2009) Distinct mechanistic activity profile of pralatrexate in comparison to other antifolates in in vitro and in vivo models of human cancers. Cancer Chemother Pharmacol 64: 993–999
Jansen G, Pieters R (1998) The role of impaired transport in (pre)clinical resistance to methotrexate: insights on new antifolates. Drug Resist Updat 1: 211–218
Marneros AG, Grossman ME, Silvers DN, Husain S, Nuovo GJ, MacGregor-Cortelli B, Neylon E, Patterson M, O’Connor OA, Zain JM (2009) Pralatrexate-induced tumor cell apoptosis in the epidermis of a patient with HTLV-1 adult T-cell lymphoma/leukemia causing skin erosions. Blood 113: 6338–6341
Mauritz R, Peters GJ, Priest DG, Assaraf YG, Drori S, Kathmann I, Noordhuis P, Bunni MA, Rosowsky A, Schornagel JH, Pinedo HM, Jansen G (2002) Multiple mechanisms of resistance to methotrexate and novel antifolates in human CCRF-CEM leukemia cells and their implications for folate homeostasis. Biochem Pharmacol 63: 105–115
O’Connor OA, Hamlin PA, Portlock C, Moskowitz CH, Noy A, Straus DJ, Macgregor-Cortelli B, Neylon E, Sarasohn D, Dumetrescu O, Mould DR, Fleischer M, Zelenetz AD, Sirotnak F, Horwitz S (2007) Pralatrexate, a novel class of antifolates with high affinity for the reduced folate carrier-type 1, produces marked complete and durable remissions in a diversity of chemotherapy refractory cases of T-cell lymphoma. Br J Haematol 139: 425–428
Odin E, Wettergren Y, Nilsson S, Willén R, Carlsson G, Spears CP, Larsson L, Gustavsson B (2003) Altered gene expression of folate enzymes in adjacent mucosa is associated with outcome of colorectal cancer patients. Clin Cancer Res 9: 6012–6019
Rothem L, Stark M, Assaraf YG (2004) Impaired CREB-1 phosphorylation in antifolate-resistant cell lines with down-regulation of the reduced folate carrier gene. Mol Pharmacol 66: 1536–1543
Sirotnak FM, DeGraw JI, Colwell WT, Piper JR (1998) A new analogue of 10-deazaaminopterin with markedly enhanced curative effects against human tumor xenografts in mice. Cancer Chemother Pharmacol 42: 313–318
Toner LE, Vrhovac R, Smith EA, Gardner J, Heaney M, Gonen M, Teruya-Feldstein J, Sirotnak F, O’Connor OA (2006) The schedule-dependent effects of the novel antifolate pralatrexate and gemcitabine are superior to methotrexate and cytarabine in models of human non-Hodgkin's lymphoma. Clin Cancer Res 12: 924–932
Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C, Niyikiza C, Paoletti P (2003) Phase III study of pemetrexed in combination with cisplatin vs cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 21: 2630–2636
Wang ES, O’Connor O, She Y, Zelenetz AD, Sirotnak FM, Moore MA (2003) Activity of a novel anti-folate (PDX, 10-propargyl 10-deazaaminopterin) against human lymphoma is superior to methotrexate and correlates with tumor RFC-1 gene expression. Leuk Lymphoma 44: 1027–1035
Acknowledgements
We acknowledge Dr Sarah MacKenzie for assistance in preparation of this manuscript. This work was supported by Allos Therapeutics Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part at the 2009 Annual Meeting of the American Association for Cancer Research.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Serova, M., Bieche, I., Sablin, MP. et al. Single agent and combination studies of pralatrexate and molecular correlates of sensitivity. Br J Cancer 104, 272–280 (2011). https://doi.org/10.1038/sj.bjc.6606063
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6606063
Keywords
This article is cited by
-
A network-based drug prioritization and combination analysis for the MEK5/ERK5 pathway in breast cancer
BioData Mining (2024)
-
Characterization of newly established Pralatrexate-resistant cell lines and the mechanisms of resistance
BMC Cancer (2021)
-
Blastic plasmacytoid dendritic cell neoplasm with response to pralatrexate
Annals of Hematology (2019)
