
Abstract
Background:
The germline BRCA2 mutation is associated with increased prostate cancer (PrCa) risk. We have assessed survival in young PrCa cases with a germline mutation in BRCA2 and investigated loss of heterozygosity at BRCA2 in their tumours.
Methods:
Two cohorts were compared: one was a group with young-onset PrCa, tested for germline BRCA2 mutations (6 of 263 cases had a germline BRAC2 mutation), and the second was a validation set consisting of a clinical set from Manchester of known BRCA2 mutuation carriers (15 cases) with PrCa. Survival data were compared with a control series of patients in a single clinic as determined by Kaplan–Meier estimates. Loss of heterozygosity was tested for in the DNA of tumour tissue of the young-onset group by typing four microsatellite markers that flanked the BRCA2 gene, followed by sequencing.
Results:
Median survival of all PrCa cases with a germline BRCA2 mutation was shorter at 4.8 years than was survival in controls at 8.5 years (P=0.002). Loss of heterozygosity was found in the majority of tumours of BRCA2 mutation carriers. Multivariate analysis confirmed that the poorer survival of PrCa in BRCA2 mutation carriers is associated with the germline BRCA2 mutation per se.
Conclusion:
BRCA2 germline mutation is an independent prognostic factor for survival in PrCa. Such patients should not be managed with active surveillance as they have more aggressive disease.
Similar content being viewed by others


Main
Prostate cancer (PrCa) is a significant public health problem. In the European Union, approximately 200 000 men are diagnosed annually with the disease. There are 35 515 cases (Cancer Research UK, 2009a) per year in the United Kingdom and 10 239 deaths (Cancer Research UK, 2009b). It is now the commonest male non-cutaneous cancer diagnosed in the United Kingdom; the lifetime risk of being diagnosed with PrCa is 1 in 10 (Cancer Research UK, 2009a). Although the increase in population screening is leading to an increase in diagnosis, many men will not develop aggressive disease. However, it is recognised that some PrCa cases have a particularly poor prognosis. Although there are some histological and stage predictors of prognosis (Kattan and Scardino, 2002), until recently, none of them have been related to inherited factors.
Multiple aetiologies have been proposed to contribute to the development of PrCa. There is strong evidence that inherited genetic factors are important and exhibit significant familial aggregation in some men, particularly when affected at a young age (Woolf, 1960; Edwards and Eeles, 2004). There is a recognised association of breast cancer with PrCa in families (Thiessen, 1974; Anderson and Badzioch, 1992; Tulinius et al, 1992). Male relatives in breast cancer families in Iceland have a 2–3-fold risk of PrCa (Sigurdsson et al, 1997). The breast cancer predisposition genes BRCA1 and BRCA2 have been reported to increase the risk of PrCa by three-fold and seven-fold, respectively, in male mutation carriers ascertained through a family history of breast cancer (Ford et al, 1994; Struewing et al, 1997; Breast Cancer Linkage Consortium, 1999). Analyses of PrCa relative risks (RR) in male mutation carriers in breast cancer families from the Breast Cancer Linkage Consortium showed an RR of 4.65 (95% CI: 3.48–6.22) of PrCa in male BRCA2 mutation carriers (the RR is 7.33 below the age of 65 years) and of 1.07 (0.75–1.54) in BRCA1 carriers (with an RR of 1.82 (1.01–3.29) for men under 65 years of age) (Thompson and Easton, 2001, 2002). The estimated cumulative incidence of PrCa by the age of 70 years is 7.5–33%. Recent studies have suggested that the risk of PrCa in BRCA2 mutation carriers may be as high as an RR of 23-fold at age 60 years (Edwards et al, 2003). Studies from Iceland have reported that germline mutations in BRCA2 may be involved not only in susceptibility to PrCa but also in the aggressiveness of the disease (Sigurdsson et al, 1997; Tryggvadottir et al, 2007). However, these individuals all carried a common founder mutation (999del5 in BRCA2). Narod et al (2008) have reported that PrCa survival in BRCA2 mutation carriers is much shorter (median survival from diagnosis was 4 years) when compared with BRCA1 carriers’ survival (median survival from diagnosis was 8 years). In PrCa, in which the BRCA2 germline mutation status was unknown, allele loss at the BRCA2 locus has been shown to be a prognostic factor for survival on univariate analysis (Edwards et al, 1998), and loss of the wild-type allele would imply a tumour suppressor mechanism in predisposition to this disease in BRCA2 mutation carriers, but it is not known whether this is a surrogate for high grade or is due to mutation per se (Knudson, 1971; Willems et al, 2008).
A BRCA2 genomic screening study has previously been undertaken by ourselves, and six potentially pathogenic germline BRCA2 mutations were found in a set of 263 PrCa patients diagnosed at ⩽55 years (2.3%) (Edwards et al, 2003). In this study we report clinical follow-up data and the results of loss of heterozygosity (LOH) analyses on PrCa tumours from the mutation carriers in this report.
We then studied a second validation data set of men with germline mutations in the BRCA2 gene from a cancer genetics clinic and assessed their survival to confirm our results in a different UK data set of male BRCA2 mutation carriers with PrCa.
Materials and methods
Patient recruitment and survival analyses
Two groups of men with PrCa were studied.
1) A series of men with PrCa from the UK Genetic Prostate Cancer Study (UKGPCS):
Patient recruitment was conducted as reported in a previous article (Eeles et al, 1997). The coding region of BRCA2 was analysed from blood DNA from 263 PrCa patients diagnosed at ⩽55 years and germline mutations in BRCA2 were found in 6 men (2.3%) (Edwards et al, 2003). The control group consisted of men from a systematic series of prostate cancer patients, age and stage matched from our prostate cancer clinic (1587 cases). The majority of patients had clinically presenting (non-screen detected) disease at diagnosis. Clinical data were collected on survival/date of last follow-up for both deleterious BRCA2 mutation carriers and controls. Supplementary Table 1 shows the demographic and clinical characteristics of prostate cancer in this group of patients, for both the cases and controls.
2) Men with PrCa who also harbour a germline BRCA2 mutation from a clinical series:
Men attending a cancer genetics clinic in Manchester, who were found on clinical genetic testing to harbour BRCA2 mutations, were reviewed from the Access clinical database and their date of death or last follow-up was ascertained from the cancer registry or from their clinical notes.
Written informed consent was obtained from individuals in this study (ethics number 06/MRE02/4).
Overall survival was measured from date of diagnosis to date of death or last follow-up. Kaplan–Meier survival analyses were undertaken with patients censored at date of last follow-up. The overall survival of those with and without germline mutations in BRCA2 in group 1 was compared using the log-rank test. The overall survival for those in group 2 was also calculated separately and in combination with group 1. The effect of other factors that could affect survival was analysed using Cox regression. The factors investigated were stage at diagnosis, incidental PSA detection, Gleason score, grade, whether they had a prostatectomy, PSA at diagnosis and age.
DNA extraction and LOH studies
Germline DNA was obtained from peripheral blood samples and extracted as reported in previous articles (Edwards et al, 1997). Tumour DNA was obtained from microdissected formalin-fixed paraffin-embedded (FFPE) blocks and extracted as reported in previous articles (Edwards et al, 1998). Normal tissue DNA was also obtained in the same way.
Four microsatellite markers, D13S260, D13S171, D13S267 and D13S1493 within and flanking the BRCA2 gene, were typed on five tumours from those men in group 1 using an ABI (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA, USA) 377 Genetic Analyser. The ‘peak height’ of the alleles was used to determine the ratio of allelic loss compared with either genomic DNA or adjacent normal tissue from paraffin blocks. Percentage allele loss for informative markers (minimum of two) was averaged for each patient.
To determine which allele was lost in the microsatellite LOH results, a sequencing method was used as described in Boettger et al (2003). We used Applied Biosystems dRhodamine chemistry on a 377 Genetic Analyser (Edwards et al, 2003). An average value for the loss was estimated by examining a number of electropherogram peak height signals in the area of the mutation.
Results
Survival analysis
The median overall survival of all BRCA2 mutation carriers was significantly shorter at 4.8 years compared with that of non-carriers at 8.5 years; log rank P=0.003 (hazard ratio 2.14 (95% CI: 1.28–3.56); see Figure 1). When analysed by method of ascertainment (see methods), the median survival of the six BRCA2 carriers in group 1 was significantly shorter at 3.6 years (P=0.002; hazard ratio 3.36 (95% CI: 1.50–7.50)), when compared with that of non-carriers. In group 2, the 15 men with germline BRCA2 mutations and PrCa had a median survival of only 5.0 years.

Kaplan–Meier survival estimates of 21 BRCA2 mutation carriers.
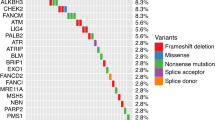
The mutations in BRCA2 in the men with PrCa are listed in Table 1.
Table 2 shows the univariate results. This shows that the following factors are associated with a poorer overall survival: germline BRCA2 mutation status, tumour (T), nodal (N) and metastasis (M) stage, tumour detected clinically rather than by PSA screening, higher Gleason score, treatment that did not involve prostatectomy, PSA at diagnosis of ⩾25 ng ml−1 and age >55 years at diagnosis. In a multivariate analysis, which is shown in Table 3, germline BRCA2 mutation status, T and nodal (N) tumour stage, higher grade, treatment that did not involve prostatectomy, PSA at diagnosis of ⩾25 ng ml−1 and higher age at diagnosis remained independent prognostic factors.
Loss of heterozygosity (LOH) results
The DNA from microdissected FFPE tumour tissue was available from 5 of 6 germline BRCA2 carriers. All five showed LOH (see Table 4). A representative microsatellite trace is shown in Figure 2.

Example of LOH seen with D13S1493 in two patients – A and D.
To estimate which allele was lost, a sequencing method was adopted. Of the five tumour samples sequenced, it was observed that patient F had lost the mutant allele, whereas patients A, D and E lost the wild-type allele. The tumour from patient B had lost the mutant allele at a low level (approx 10%; Table 4). Representative sequence traces are shown in Figure 3.

Examples of allele loss in patients A, D and F. Arrows show the position of a representative peak for signal analysis.
Discussion
We have shown that men with PrCa, who also harbour a deleterious germline mutation in the BRCA2 gene, have a poorer overall survival. This has been shown in a small sample of men who were diagnosed at ⩽55 years with PrCa, compared with those diagnosed at a similarly young age, but who did not harbour a germline mutation as determined by coding sequence analysis from a previous study, and also men from a systematic series of prostate cancer cases in one centre: group 1 (Edwards et al, 2003). We have validated this in a separate data set of men who have had PrCa at any age and who have been found to have a germline mutation in BRCA2 by a clinical genetic testing service, in which genetic testing was offered as part of genetic counselling of families with mutations: group 2. These are usually men within breast cancer families in which women have initially been tested to determine the cause of familial breast cancer clustering within the family. Again, they have a poorer prognosis than men who do not harbour a germline BRCA2 mutation.
It has been shown that local extent or T stage, N stage and presence or not of M stage and higher PSA at presentation are all predictors of poorer survival (Kattan and Scardino, 2002). It was therefore very important to determine whether the poorer survival associated with the presence of a germline BRCA2 mutation was independent. The multivariate analysis confirms that the presence of a germline BRCA2 mutation is a marker of poorer overall survival per se.
This has implications for the detection and management of men with PrCa who are found to harbour germline mutations in the BRCA2 gene, as their poorer survival would be a contraindication for active surveillance. It is not yet known whether these men should have a particular modality of treatment (e.g., surgery rather than radiation), as the sample sizes in this paper are too small and treatment data are incomplete in these data sets to be able to determine this.
Before 2008, the only data available on survival in men with PrCa who harbour germline mutations in BRCA2 were from Iceland, where there is a founder mutation (Thorlacius et al, 1996; Sigurdsson et al, 1997; Tryggvadottir et al, 2007). Tryggvadottir et al (2007) found that PrCa carriers with the BRCA2 999del5 mutation had a lower mean age at diagnosis, more advanced PrCa as assessed by stage and grade, and a shorter median survival time compared with non-carriers. Their study showed a median survival time for carriers (30) of 2.1 years, which was significantly shorter than the 12.4 years for non-carriers (497). The survival time of the Icelandic carriers is much shorter than that reported for our early-onset group 1 carriers of 4 years. One factor to be noted in the Tryggvadottir et al (2007) study is that all of the 527 PrCa patients were related to breast cancer patients, 28% of whom were first-degree relatives. Of the 527 patients, 30 were determined to be 999del5 carriers. It is therefore possible that the poorer prognosis that was reported could have pertained only to this specific mutation type. This result, and its potential specificity by virtue of mutation and population, has been discussed further by Boormans and Schröder (2007). In contrast, we have shown that in the UK population, men with a variety of other mutations in BRCA2, are likely to have a similarly poorer survival, and therefore the poorer survival is likely to be related to different deleterious mutations in the BRCA2 gene.
We found LOH in the five available PrCa tumours from men in group 1.Three of the five tumours had lost the wild-type allele. This is consistent with a tumour suppressor model and indicative of a causal relationship between BRCA2 germline mutations and predisposition to PrCa in these individuals. Loss of the mutant allele was also observed. The implication is that for disease causation, maybe a gene dosage effect is important.
Tommiska et al (2008) have recently proposed a model of ‘conditional haploinsufficiency’, whereby defects in genes such as BRCA1 or BRCA2 can disrupt the regulation of other important genome integrity monitoring genes such as ATM. In this hypothesis, the local effects of these predisposing genes, by virtue of perhaps increased DNA double-strand breaks, would ultimately cause ATM protein inactivation as the cells progressed to malignancy. Although this haploinsufficiency mechanism has only been investigated in breast cancer, it may have implications for the development of PrCa in the carriers that we studied, and could explain why we saw loss of mutant alleles in comparison with the classical loss of wild type.
There is very scant literature on the ‘classical’ loss of wild-type alleles from PrCa patients who are BRCA2 carriers. Gudmundsson et al (1995) and Grönberg et al (2001) have reported LOH in PrCa patients. Gudmundsson et al (1995) investigated five high-risk breast cancer families and found seven men with PrCa, six of whom had LOH at the BRCA2 locus. Grönberg et al (2001) studied a breast/prostate family (three breast cancer, five PrCa) that was found to have a deleterious BRCA2 mutation (6051delA). Of the four brothers with PrCa, two had LOH (loss of WT) and two retained heterozygosity.
Willems et al (2008) reported on the screening of a large series of kConFab Australian BRCA2 breast cancer families. There were many men with PrCa in these families; however, 20 were confirmed to be BRCA2 carriers, and 14 of them had tumours that were available for analysis by multiplex ligation-dependent probe amplification (MLPA). This technique is able to assay the entire BRCA2 gene for loss of promoter and coding regions. Of the 14 BRCA2 carriers, 10 showed loss of heterozygosity by the MLPA method. The set comprised six men with a known family history of PrCa and all the six showed LOH at BRCA2. The conclusion was that the wild-type allele was most often lost, but for the four cases that showed no LOH, epigenetic and haploinsufficiency models were postulated as possible mechanisms. This allele determination was assessed by a sequencing technique that is similar to the one we used. Willems et al (2008) reported comprehensive clinical data on their carriers and it is noteworthy that all 10 PrCa cases presented with high Gleason scores of 9. This is a finding that is similar to the high-grade presentation of the men in our study group 1. The risk estimates of 3.5-fold for PrCa, as reported by Willems et al (2008), are lower than the RR reported by us in 2003 (Edwards et al, 2003).
In 2008, Narod et al (2008) reported data on a panel of PrCa patients with BRCA1 and BRCA2 germline mutations identified from breast cancer families. For the combined group of known and inferred carriers, the median survival for 183 BRCA2 patients was 4.0 years vs 8.0 years for the 119 men in the BRCA1 group. When only known carriers were analysed, the results were 5.0 years (67 BRCA2 known carriers) and 15.0 years (37 BRCA1 known carriers). Although there is a difference in the survival of BRCA1 patients, which is discussed by the authors, the poorer survival of BRCA2-related patients is not in dispute. Use of either survival measure, 4 or 5 years, illustrates that the median survival of the patients studied is very similar to that observed in our study of 4.8 years.
It is not yet known whether earlier detection of PrCa in men with germline mutations in BRCA2 will result in a better outcome and whether PSA screening is suitable for such a population. The IMPACT study (Identification of Men with a genetic predisposition to ProstAte Cancer: Targeted screening in BRCA1 and BRCA2 mutation carriers and controls) has been developed to investigate the role of targeted PrCa screening in male BRCA1 and BRCA2 gene mutation carriers using an annual PSA screen (Mitra et al, 2007). Early data have suggested that men will uptake screening and that PrCa was twice as likely in BRCA1/2 mutation carriers (Horsburgh et al, 2005).
Our data have shown that the observation in Iceland, which shows that men who carry the Icelandic founder mutation in the BRCA2 gene who also develop PrCa have a poorer survival, is not restricted to this mutation and we have shown that this is due to the presence of a deleterious mutation within the BRCA2 gene per se. There is some dispute about the precise frequency of germline BRCA2 mutation in men with PrCa. This is reported to be about 1% in men aged ⩽55 years in a US series (Agalliu et al, 2007), but this may be equivalent to 65–68 years at clinical diagnosis, as the prevalence of PSA screen-detected disease is higher in the United States. This is likely to be the case, as in this US series, 67.3% of men had a PSA of <10. We have found that 2.3% of men diagnosed with clinically presenting disease (non-PSA detected) have a germline BRCA2 mutation (Edwards et al, 2003). Even if the incidence of a germline mutation in men with young-onset (defined as ⩽55 years at diagnosis) PrCa was as low as 2%, the finding of a BRCA2 mutation would be an indication to avoid active surveillance in these patients, as they have a more aggressive disease outcome.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agalliu I, Karlines E, Kwon EM, Iwasaki LM, Diamond A, Ostrander EA, Stanford JL (2007) Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. Br J Cancer 97: 826–831
Anderson DE, Badzioch MD (1992) Breast cancer risks in relatives of male breast cancer patients. J Natl Cancer Inst 84: 1114–1117
Boettger MB, Sergi C, Meyer P (2003) BRCA1/2 mutation screening and LOH analysis of lung adenocarcinoma tissue in a multiple-cancer patient with a strong family history of breast cancer. J Carcinog 2: 5, doi: 10.1186/1477-3163-2-5
Boormans JL, Schröder FH (2007) Re: Prostate cancer progression and survival in BRCA2 mutation carriers. Eur Urol 52: 1529
Breast Cancer Linkage Consortium (1999) Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91: 1310–1316
Cancer Research UK (2009a) Prostate cancer – UK incidence statistics. Available: http://info.cancerresearchuk.org/cancerstats/types/prostate/incidence/ Last accessed 18 January 2010
Cancer Research UK (2009b) Prostate cancer – UK mortality statistics. Available: http://info.cancerresearchuk.org/cancerstats/types/prostate/mortality/?a=5441 Last accessed 18 January 2010
Edwards SM, Dearnaley DP, Ardern-Jones A, Hamoudi RA, Easton DF, Ford D, Shearer R, Dowe A, Eeles RA (1997) No germline mutations in the dimerization domain of MXI1 in prostate cancer clusters. The CRC/BPG UK Familial Prostate Cancer Study Collaborators. Cancer Research Campaign/British Prostate Group. Br J Cancer 76: 992–1000
Edwards SM, Dunsmuir WD, Gillett CE, Lakhani SR, Corbishley C, Young M, Kirby RS, Dearnaley DP, Dowe A, Ardern-Jones A, Kelly J, Spurr N, Barnes DM, Eeles RA (1998) Immunohistochemical expression of BRCA2 protein and allelic loss at the BRCA2 locus in prostate cancer. CRC/BPG UK Familial Prostate Cancer Study Collaborators. Int J Cancer 78: 1–7
Edwards SM, Eeles RA (2004) Unravelling the genetics of prostate cancer. Am J Med Genet C Semin Med Genet 129C: 65–73
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, Dearnaley DP, Ardern-Jones A, Murkin A, Dowe A, Kelly J, Williams S, Oram R, Stevens M, Teare DM, Ponder BA, Gayther SA, Easton DF, Eeles RA (2003) Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 72: 1–12
Eeles RA, Dearnaley DP, Ardern-Jones A, Shearer RJ, Easton DF, Ford D, Edwards S, Dowe A (1997) Familial prostate cancer: the evidence and the cancer research campaign/British prostate group (CRC/BPG) UK familial prostate cancer study. Br J Urol 79 (Suppl 1): 8–14
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE (1994) Risks of cancer in BRCA1-mutation carriers. The breast cancer linkage consortium. Lancet 343: 692–695
Grönberg H, Ahman AK, Emanuelsson M, Bergh A, Damber JE, Borg A (2001) BRCA2 mutation in a family with hereditary prostate cancer. Genes Chromosomes Cancer 30: 299–301
Gudmundsson J, Johannesdottir G, Bergthorsson JT, Arason A, Ingvarsson S, Egilsson V, Barkardottir RB (1995) Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12-q13. Cancer Res 55: 4830–4832
Horsburgh S, Matthew A, Bristow R, Trachtenberg J (2005) Male BRCA1 and BRCA2 mutation carriers: a pilot study investigating medical characteristics of patients participating in a prostate cancer prevention clinic. Prostate 65: 124–129
Kattan MW, Scardino PT (2002) Prediction of progression: nomograms of clinical utility. Clin Prostate Cancer 1: 90–96
Knudson Jr AG (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68: 820–823
Mitra AV, Bancroft EK, Eeles RA (2007) IMPACT Steering Committee and Collaborators. A review of targeted screening for prostate cancer: introducing the IMPACT study. BJU Int 99: 1350–1355
Narod SA, Neuhausen S, Vichodez G, Armel S, Lynch HT, Ghadirian P, Cummings S, Olopade O, Stoppa-Lyonnet D, Couch F, Wagner T, Warner E, Foulkes WD, Saal H, Weitzel J, Tulman A, Poll A, Nam R, Sun P (2008) Rapid progression of prostate cancer in men with a BRCA2 mutation. Br J Cancer 99: 371–374
Sigurdsson S, Thorlacius S, Tomasson J, Tryggvadottir L, Benediktsdottir K, Eyfjörd JE, Jonsson E (1997) BRCA2 mutation in Icelandic prostate cancer patients. J Mol Med 75 (10): 758–761
Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA (1997) The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336: 1401–1408
Thiessen E (1974) Concerning a familial association between breast cancer and both prostatic and uterine malignancies. Cancer 34: 1102–1107
Thompson D, Easton D, Breast Cancer Linkage Consortium (2001) Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet 68: 410–419
Thompson D, Easton DF, Breast Cancer Linkage Consortium (2002) Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94: 1358–1365
Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, Tulinius H, Ogmundsdottir HM, Eyfjörd JE (1996) A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat Genet 13: 117–119
Tommiska J, Bartkova J, Heinonen M, Hautala L, Kilpivaara O, Eerola H, Aittomäki K, Hofstetter B, Lukas J, von Smitten K, Blomqvist C, Ristimäki A, Heikkilä P, Bartek J, Nevanlinna H (2008) The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene 27: 2501–2506
Tryggvadottir L, Vidarsdottir L, Thorgeirsson T, Jonasson JG, Olafsdottir EJ, Olafsdottir GH, Rafnar T, Thorlacius S, Jonsson E, Eyfjord JE, Tulinius H (2007) Prostate cancer progression and survival in BRCA2 mutation carriers. J Natl Cancer Inst 99: 929–935
Tulinius H, Egilsson V, Olafsdottir GH, Sigvaldason H. (1992) Risk of prostate, ovarian, and endometrial cancer among relatives of women with breast cancer. BMJ 305: 855–857
Willems AJ, Dawson SJ, Samaratunga H, De Luca A, Antill YC, Hopper JL, Thorne HJ, and kConFab Investigators (2008) Loss of Heterozygosity at the BRCA2 Locus Detected by Multiplex Ligation-Dependent Probe Amplification is Common in Prostate Cancers from Men with a Germline BRCA2 Mutation. Clin Cancer Res 14: 2953–2961
Woolf CM (1960) An investigation of the familial aspects of carcinoma of the prostate. Cancer 13: 739–744
Acknowledgements
We thank all the patients and their families for participating in this study. We thank S Clinton for cutting the slides, and Noemi Garcia, Sarah Jugurnauth and Sandra Edwards for helpful discussions. This work was supported by Cancer Research UK grant no C5047/A3354, The Prostate Cancer Research Foundation, The Institute of Cancer Research, The Everyman Campaign and The Genesis Charity. The funds for the ABI 377 used in this study were generously provided by the legacy of the late Marion Silcock. We acknowledge support from the NHS to The Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Edwards, S., Evans, D., Hope, Q. et al. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br J Cancer 103, 918–924 (2010). https://doi.org/10.1038/sj.bjc.6605822
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605822
Keywords
This article is cited by
-
Implications of targeted next-generation sequencing for bladder cancer: report of four cases
Journal of Genetic Engineering and Biotechnology (2021)
-
Genetik des Prostatakarzinoms
Journal für Urologie und Urogynäkologie/Österreich (2019)
-
The novel long noncoding RNA u50535 promotes colorectal cancer growth and metastasis by regulating CCL20
Cell Death & Disease (2018)
-
Prostate-specific antigen velocity in a prospective prostate cancer screening study of men with genetic predisposition
British Journal of Cancer (2018)
-
Mutations in BRCA2 and taxane resistance in prostate cancer
Scientific Reports (2017)
