
Abstract
Background:
Recent studies have indicated that prostate cancer patients with the TMPRSS2–ERG gene fusion have a higher risk of recurrence. To identify markers associated with TMPRSS2–ERG fusion and prognostic of biochemical recurrence, we analysed a cohort of 139 men with prostate cancer for 502 molecular markers.
Methods:
RNA from radical prostatectomy tumour specimens was analysed using cDNA-mediated, annealing, selection, extension and ligation (DASL) to determine mRNAs associated with TMPRSS2–ERG T1/E4 fusion and prognostic of biochemical recurrence. Differentially expressed mRNAs in T1/E4-positive tumours were determined using significance analysis of microarrays (false discovery rate (FDR) <5%). Univariate and multivariate Cox regression determined genes, gene signatures and clinical factors prognostic of recurrence (P-value <0.05, log–rank test). Analysis of two prostate microarray studies (GSE1065 and GSE8402) validated the findings.
Results:
In the 139 patients from this study and from a 455-patient Swedish cohort, 15 genes in common were differentially regulated in T1/E4 fusion-positive tumours (FDR <0.05). The most significant mRNAs in both cohorts coded ERG. Nine genes were found prognostic of recurrence in this study and in a 596-patient Minnesota cohort. A molecular recurrence score was significant in prognosticating recurrence (P-value 0.000167) and remained significant in multivariate analysis of a mixed clinical model considering Gleason score and TMPRSS2–ERG fusion status.
Conclusions:
TMPRSS2–ERG T1/E4 fusion-positive tumours had differentially regulated mRNAs observed in multiple studies, the most significant one coded for ERG. Several mRNAs were consistently associated with biochemical recurrence and have potential clinical utility but will require further validation for successful translation.
Similar content being viewed by others

Main
Prostate cancer prognosis after radical prostatectomy is incompletely assessed by clinical markers such as Gleason score, tumour, node, metastasis (TNM) stage, surgical margin status and preoperative prostate-specific antigen (PSA) level. Recently identified genetic markers could provide clinical utility for prostate cancer progression and recurrence independent of the current clinical markers, thereby improving patient management. Examples of such markers include the E twenty-six (ETS) family of transcription factors that were identified through outlier profile analysis that detected an elevated expression of ETS factors (Tomlins et al, 2005). Normal and benign prostate tissues, as well as prostatic intraepithelial neoplasia lesions, lack the expression of ERG, whereas expression of TMPRSS2–ERG fusion mRNAs occurs uniquely in prostate adenocarcinoma because of the fusion of a promoter/enhancer region of an androgen-responsive prostate-specific serine protease 2-encoding gene, TMPRSS2, to the v-ets erythroblastosis virus E26 oncogene homologue, ERG, or the ETS variant 1 gene, ETV1 (Tomlins et al, 2005; Narod et al, 2008), and over 20 other gene fusion variants (Perner et al, 2006; Mehra et al, 2007; Tu et al, 2007). Fusion of TMPRSS2 (21q22.2) and ERG (21q22.3), which are within 3 Mb of each other, results from a chromosome 21 microdeletion in approximately two-thirds of fusion cases (Yoshimoto et al, 2006b). The most common variant is a recombination between exon 1 of TMPRSS2 and exon 4 of ERG, designated T1/E4 (Clark et al, 2007; Jhavar et al, 2008), and some studies indicate that this variant accounts for 85% of reported fusions (Wang et al, 2006; Mehra et al, 2007).
Both the prevalence and prognostic significance of the TMPRSS2–ERG T1/E4 fusion have been examined in multiple studies with some discrepancy in results. Although TMPRSS2–ERG fusion has typically been reported as prevalent in 40–50% of prostate tumours, the range has varied by as much as 25–60% (Nam et al, 2007; Setlur et al, 2008; Sun et al, 2008; Yoshimoto et al, 2008; Hofer et al, 2009; Mosquera et al, 2009). The techniques used for TMPRSS2–ERG detection, novel potential fusion products and genetic differences in population cohorts may account for these discrepancies. This is highlighted by the finding of Mosquera et al (2009) that Caucasian populations were less likely to have the TMPRSS2–ERG fusion and this could explain the 22.6% prevalence rate observed by Setlur et al (2008) in a Swedish cohort of 455 prostate cancer cases. Irrespective of the cause for reported variation in prevalence, further studies are needed to clearly define the prevalence rate of TMPRSS2–ERG fusions in the context of population genetics.
Commensurate with the range in prevalence of TMPRSS2–ERG fusions is the variation in reported prognostic significance. Several studies have indicated that TMPRSS2–ERG fusions confer a worse prognosis (Nam et al, 2007; Attard et al, 2008a; Clark et al, 2008), whereas others have not (Gopalan et al, 2009). Discrepancies in the reported prognostic significance of TMPRSS2–ERG fusions can be explained in a similar manner by factors affecting the variation in prevalence of TMPRSS2–ERG (i.e., cohort race/ethnicity, fusion detection technique), and are also liable to the primary end point of the study (i.e., biochemical recurrence, overall survival). The complexity contributing to the discordant prognostic significance of TMPRSS2–ERG fusions was mentioned by FitzGerald et al (2008), who reported that TMPRSS2–ERG fusions did not result in reduced survival, but that the combination of TMPRSS2–ERG fusion and amplification of the fusion gene conferred a worse prognosis. As in the case of TMPRSS2–ERG fusion prevalence, to properly understand the prognostic significance of TMPRSS2–ERG fusions, further standardised studies are needed. Proper patient tracking and characterisation in combination with proper sample management can ensure accurate molecular characterisation and spur translation of clinical tools.
In an effort to better characterise the molecular implications of TMPRSS2–ERG fusions in prostate cancer and to identify other genetic prognostic markers, we have utilised RNA extracted from radical prostatectomy specimens in a prospective cohort previously characterised for TMPRSS2–ERG T1/E4 fusion transcript expression by qualitative PCR (Nam et al, 2007). This cohort (hereafter referred to as the Toronto cohort) of 165 patients had 81 (49%) fusion-positive tumours in which patients with a detectable fusion transcript had a significantly higher risk of biochemical recurrence (58% at 5 years) than did patients whose tumours did not express detectable fusion transcripts (8% at 5 years). Therefore, TMPRSS2–ERG T1/E4 fusion was found to be a strong prognostic factor independent of grade, stage and PSA level in this cohort (Nam et al, 2007). We have subsequently made use of RNA from 139 of the 165 patients and characterised the expression of 502 cancer-related genes using the cDNA-mediated, annealing, selection, extension and ligation (DASL) assay.
Materials and methods
RNA samples
Total RNA samples from frozen prostate tumour specimens used in this study were prepared previously (Nam et al, 2007). Aliquoted RNA samples were shipped on dry ice to the Emory Biomarker Service Center (EBSC), Emory University, Atlanta, GA, USA, for use in the DASL assay. RNA concentration was quantified by a Nanodrop spectrophotometer (Wilmington, DE, USA) and quality was assessed using the Agilent Bioanalyzer (Foster City, CA, USA) for which RNA integrity number of >7 was used as a quality criteria.
DASL assay performance, reproducibility and data normalisation
The DASL assay was performed on Illumina's 502-gene Human Cancer Panel using 200 ng of input RNA. The manufacturer's instructions were followed without any changes. Samples were hybridised on two Universal Array Matrices (UAMs). Hybridised UAMs were scanned using the BeadStation 500 Instrument (Illumina, San Diego, CA, USA). Data were interpreted and quantile normalised using GenomeStudio v1.0.2 (Illumina). Experimental replicates (same RNA assayed twice) were assessed for reproducibility and subsequently averaged so as to represent each patient's tumour sample with one gene expression profile.
Data analysis and meta-analysis
Differential mRNA expression of TMPRSS2–ERG T1/E4 fusion-positive vs fusion-negative tumours was assessed using significance analysis of microarrays (Tusher et al, 2001) for which 1000 random class assignment permutations estimated a false discovery rate (FDR) ⩽5%. Hierarchical clustering was generated in R using the heatmap.2 package in which distance was computed using a Euclidean dissimilarity metric with an average-linkage clustering algorithm. Data were displayed with mRNA intensity z-score normalised. Gene Ontology analysis was conducted using the R package GOstats with a significance value of P<0.01 of over-representation computed by the hypergeometric test using the lumiHumanAll.db annotation file. Univariate Cox proportional hazards regression was conducted in R using the Cox proportional hazards survival package (CoxPH) and was conducted on each gene expression profile and clinical factor independently. Multivariate Cox analysis considered the clinical factors that were significant (P<0.05) in univariate analysis, as well as a recurrence predictor built as a weighted average of the expression level of genes, which were significant in univariate analysis in both the Toronto data set and that from Nakagawa et al (2008). Kaplan–Meier curves were generated in R using the survival package, and significance testing utilised the survdiff function for which the log–rank test determined the P-value. Analysis utilised expression profiles from studies by both Setlur et al (2008) and Nakagawa et al (2008), which were downloaded from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and had the series numbers GSE8402 and GSE10645, respectively. The same differential annotation and prognostic analyses methods described above were used on the meta-analysis sets.
Results
After RNA and assay quality control, 139 patient tumours were characterised on the DASL assay for 502 cancer-related genes (GEO series GSE18655). Seven samples were run as experimental replicates to estimate assay reproducibility for which an average Pearson's R2 of 0.965 indicated highly reproducible data (Supplementary Figure 1). Moreover, unsupervised hierarchical clustering of all samples and probes resulted in experimental replicates clustering together without exception (Supplementary Figure 2). The Toronto cohort, a subset of that previously characterised for clinical markers (Nam et al, 2007), includes 69 patients with TMPRSS2–ERG T1/E4 fusion-positive tumours and 70 prostate tumours that were TMPRSS2–ERG fusion negative. Fusion status indicated a significantly worse outcome with respect to biochemical recurrence (Figure 1A, P-value 3.54 × 10−8 log–rank test) similar to that observed in the entire cohort (Nam et al, 2007). As previously reported, patients with TMPRSS2–ERG fusion-positive tumours had a significantly higher expression of ERG transcripts (Figure 1B, P-value 3.48 × 10−11, two-sided Student's t-test), which is most likely a result of androgen-responsive promoter elements in TMPRSS2 driving expression (Tomlins et al, 2005). Overexpression of ERG was validated using reverse transcriptase PCR, which corroborated the ERG overexpression found by microarray results (Supplementary Figure 3, P-value 8.13 × 10−10, two-sided Student's t-test).

Characteristics of prostate cancer patients with and without the T1/E4 TMPRSS2–ERG fusion. (A) Patients with TMPRSS2–ERG fusion-positive tumours experienced a higher rate of biochemical recurrence opposed to those who did not have the gene fusion (P-value 3.54 × 10−8, log–rank test). (B) ERG expression was upregulated in TMPRSS2–ERG fusion-positive tumours by 3.07-fold (P-value 3.48 × 10−11, Student's t-test).
To investigate molecular biomarkers differentially regulated in TMPRSS2–ERG fusion-positive tumours, significance testing was conducted using significance analysis of microarrays (Tusher et al, 2001) for both the Toronto cohort and that of the 455-patient Swedish cohort (Setlur et al, 2008). Using an FDR ⩽5% yielded 51 genes differentially regulated in TMPRSS2–ERG fusion-positive tumours in the Toronto cohort (Supplementary Table 1). Nine upregulated genes and six downregulated genes were validated by replicating the analysis on the Swedish cohort (Setlur et al, 2008), which was characterised for expression of 6144 transcripts (Figure 2A, FDR <5%). In both the Toronto and Swedish cohorts, ERG was uniquely the most significant differentially regulated transcript in TMPRSS2–ERG fusion-positive tumours (Supplementary Figure 4). Genes annotated for mismatch base repair and histone deacetylation functions were over-represented in Gene Ontology analysis of common upregulated genes in T1/E4-positive tumours (Supplementary Table 2, P<0.01). Downregulated genes were over-represented for annotations that included the insulin-like growth factor (IGF) and Jak-Stat signalling pathways, suggesting that these pathways may be attenuated in T1/E4-positive tumours (Supplementary Table 2, P<0.01). Hierarchical clustering of tumour expression profiles across common differentially regulated genes resulted in segregation of TMPRSS2–ERG fusion-positive tumours (Figures 2B and C), suggesting that TMPRSS2–ERG fusion-positive tumours have a distinct molecular metabolism that is replicated in multiple cohorts.

Validated genes differentially expressed in TMPRSS2–ERG fusion-positive tumours. (A) Significance testing of genes differentially regulated in TMPRSS2–ERG fusion-positive prostate tumours in the Toronto cohort of 139 patients characterised for 502 genes (solid black line) was validated in a Swedish cohort (Setlur et al, 2008) of 455 patients characterised for 6144 genes (dashed black line). Nine genes upregulated with the TMPRSS2–ERG fusion in both cohorts are shown at the top (red box), whereas six genes downregulated in both cohorts are shown at the bottom (green box). Hierarchical clustering of the 15 common differentially expressed genes segregates TMPRSS2–ERG fusion-positive tumours as indicated in black above the heatmaps and below the clustering dendrogram in (B) the Toronto cohort of 139 patients and (C) the Swedish cohort of 455 patients.
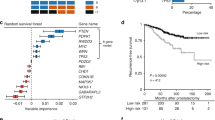
To determine the molecular factors associated with biochemical recurrence (defined as a PSA increase of ⩾0.2 ng ml−1 on at least two consecutive measurements that are at least 3 months apart), univariate Cox proportional hazards regression was conducted in the Toronto cohort and replicated in a 596-patient Minnesota cohort (Nakagawa et al, 2008). The Toronto data set yielded 16 genes associated with recurrence and 11 genes associated with non-recurrence (Supplementary Table 3, P<0.05). Repeating this analysis in the Minnesota cohort validated five genes associated with biochemical recurrence and four genes associated with non-recurrence (Figure 3A, P<0.05). Gene Ontology functional annotation of genes commonly associated with recurrence yielded over-representation of deoxyribosylthymine monophosphate biosynthesis, negative regulation of leukocyte activation – specifically T- and B-cell lymphocytes – and inhibition of cell-matrix adhesion. Conversely, annotation of genes associated with non-recurrence resulted in cell-matrix adhesion and collagen binding (Supplementary Table 4, P<0.01). Common genes prognostic of recurrence were used to build a recurrence score calculated as the sum product of the expression intensity of each gene by its Cox coefficient determined by regression analysis. Ordering samples by the recurrence score in a supervised heatmap produced a trend whereby patients who did not have recurrence were separated from those who did in both the Toronto and Swedish cohorts (Figures 3B and C). More importantly, the recurrence score was significant in univariate Cox regression and remained significant in a multivariate model considering clinical factors that were significant (P<0.05) in the univariate analysis, namely, preoperative PSA level, Gleason score and TMPRSS2–ERG fusion status (Table 1 – Toronto cohort). Furthermore, the nine-gene expression recurrence score was significantly associated with biochemical recurrence by itself (Figure 4A, P-value 0.000167) and in a multivariate model considering the Gleason score and TMPRSS2–ERG fusion status (i.e., those clinical data significant in univariate analysis; Figure 4B, P-value 4.15 × 10−7).

Common genes prognostic of biochemical recurrence. (A) Univariate Cox proportional hazards regression determined genes associated with biochemical recurrence in the Toronto cohort of 139 patients, and a Minnesota cohort of 596 patients (Nakagawa et al, 2008) identified seven genes in common, five associated with recurrence and two associated with non-recurrence. Supervised heatmaps ordered by the seven-gene expression recurrence score showed an increased incidence of recurrence with increased recurrence score in (B) the Toronto cohort of 139 patients and (C) the Minnesota cohort of 596 patients (Nakagawa et al, 2008).

Kaplan–Meier survival analysis of the Toronto cohort. (A) Seven-gene expression recurrence score used to segregate patients into good and poor prognostic categories. (B) A mixed clinical model composed of the Gleason score, TMPRSS2–ERG fusion status and the seven-gene expression recurrence score is better able to prognosticate recurrence.
Discussion
The previously identified TMPRSS2–ERG T1/E4 gene fusion represents a potentially important factor in prostate cancer progression, the prognostic value of which has yet to be fully defined. In this cohort, the TMPRSS2–ERG T1/E4 gene fusion is highly associated with biochemical recurrence and these findings have been supported by other studies (Clark et al, 2008; Attard et al, 2008b), although some have not found a worse prognosis conferred by fusion status (Gopalan et al, 2009). Also of primary clinical importance, and even less defined, is the potential predictive value of TMPRSS2–ERG fusion (i.e., is treatment course predicted by fusion status?). To these important prognostic and predictive end points, this study makes two significant contributions.
First, this study finds 27 genes prognostic of biochemical recurrence, with 16 genes associated with recurrence and 11 genes associated with non-recurrence. Replication of this analysis in a Minnesota cohort of 596 patients (Nakagawa et al, 2008) validated five genes associated with recurrence and four associated with non-recurrence (Figure 3A). Use of these nine genes to create a recurrence score yielded a highly significant prognostic variable that could be used to segregate patients into high- and low-risk categories. This nine-gene expression recurrence score remained significant in multivariate analysis considering clinical factors such as Gleason score and other genetic factors such as TMPRSS2–ERG fusion status. Although translation of any such prognostic test would require more analysis and validation, not only by reverse transcriptase PCR but also in other independent cohorts, this study contributes to the current body of knowledge with respect to identifying biomarkers with the potential to improve prostate cancer patient management. Furthermore, certain genes that we found prognostic of recurrence, which were not validated in the Minnesota cohort, have previously been linked to prostate cancer progression, such as mucin 1 (Cozzi et al, 2005), which has also been proposed as a candidate for targeted therapy (Li and Cozzi, 2007), and may warrant further consideration as a prognostic biomarker.
Second, this study finds 51 differentially expressed genes dependent on TMPRSS2–ERG T1/E4 fusion status, 15 (Supplementary Table 1, in bold) of which are validated in a 455-patient Swedish cohort (Setlur et al, 2008) using the same analysis. Functional annotation of the 15 genes replicated in both cohorts suggests that TMPRSS2–ERG fusion is associated with mismatch base repair and histone deacetylation molecular functions. It is therefore a reasonable hypothesis that therapeutic modalities targeting these pathways may be well suited for TMPRSS2–ERG fusion-positive prostate cancer patients. The histone deacetylation inhibitors SAHA (vorinostat) and LBH589, both currently being tested in clinical trials, can inhibit androgen receptor (AR)-mediated transcriptional activation of a number of genes including TMPRSS2–ETS fusion genes (Welsbie et al, 2009). Similarly, mismatch base repair has the potential to be targeted with the use of poly(ADP-ribose) polymerase inhibitors, a class of compounds showing significant potential in the treatment of hereditary (BRCA mutation) breast cancer and currently being tested in several clinical trials (Fong et al, 2009). Conversely, prostate cancer patients without the TMPRSS2–ERG fusion gene may be better suited for therapies that target IGFs and Jak-Stat signalling, in which IGF signalling can be targeted through the use of tyrosine kinase inhibitors or monoclonal antibodies. Finally, although not replicated in the Swedish cohort, we found attenuation of the potent tumour-suppressor phosphatase and tensin homologue, PTEN, in TMPRSS2–ERG fusion-positive tumours. Loss of PTEN expression has been strongly associated with prostate cancer progression (Koksal et al, 2004; Bertram et al, 2006; Yoshimoto et al, 2006a; Schmitz et al, 2007) and has recently been implicated as cooperating with TMPRSS2–ERG fusion in high-grade prostatic intraepithelial neoplasia (Carver et al, 2009; King et al, 2009). Loss of PTEN results in PI3K activation, which in turn can be targeted with emerging modalities inhibiting PI3K or PI3K and mTor such as SF1126 and BEZ235, respectively. This study supports other evidence that TMPRSS2–ERG fusion status may have important predictive implications of such therapies in the efficacious treatment of prostate cancer.
Our findings of several replicated mRNAs prognostic of recurrence indicate that there are consistent hallmarks of refractory disease that may have clinical utility. Furthermore, the TMPRSS2–ERG fusion may have an important prognostic and predictive value, because of which differentially regulated mRNAs (most notably ERG) in repeated experiments suggest that targeted therapy in prostate cancer could be aided by this biomarker. These hypotheses will require standardised clinical trials with proper patient tracking and sample handling to translate these molecular targets into meaningful clinical tools.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL, Moller H, Reuter V, De Bono JS, Scardino P, Cuzick J, Cooper CS (2008a) Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 27 (3): 253–263
Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL, Moller H, Reuter V, De Bono JS, Scardino P, Cuzick J, Cooper CS (2008b) Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 27 (3): 253–263
Bertram J, Peacock JW, Fazli L, Mui AL, Chung SW, Cox ME, Monia B, Gleave ME, Ong CJ (2006) Loss of PTEN is associated with progression to androgen independence. Prostate 66 (9): 895–902
Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP (2009) Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 41 (5): 619–624
Clark J, Merson S, Jhavar S, Flohr P, Edwards S, Foster CS, Eeles R, Martin FL, Phillips DH, Crundwell M, Christmas T, Thompson A, Fisher C, Kovacs G, Cooper CS (2007) Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene 26 (18): 2667–2673
Clark JP, Munson KW, Gu JW, Lamparska-Kupsik K, Chan KG, Yoshida JS, Kawachi MH, Crocitto LE, Wilson TG, Feng Z, Smith SS (2008) Performance of a single assay for both type III and type VI TMPRSS2: ERG fusions in noninvasive prediction of prostate biopsy outcome. Clin Chem 54 (12): 2007–2017
Cozzi PJ, Wang J, Delprado W, Perkins AC, Allen BJ, Russell PJ, Li Y (2005) MUC1, MUC2, MUC4, MUC5AC and MUC6 expression in the progression of prostate cancer. Clin Exp Metastasis 22 (7): 565–573
FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, Hurtado-Coll A, Fazli L, Rajput AB, Gleave ME, Cox ME, Ostrander EA, Stanford JL, Huntsman DG (2008) Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC Cancer 8: 230
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, De Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361 (2): 123–134
Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, Fine SW, Eastham JA, Scardino PT, Scher HI, Tickoo SK, Reuter VE, Gerald WL (2009) TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 69 (4): 1400–1406
Hofer MD, Kuefer R, Maier C, Herkommer K, Perner S, Demichelis F, Paiss T, Vogel W, Rubin MA, Hoegel J (2009) Genome-wide linkage analysis of TMPRSS2-ERG fusion in familial prostate cancer. Cancer Res 69 (2): 640–646
Jhavar S, Reid A, Clark J, Kote-Jarai Z, Christmas T, Thompson A, Woodhouse C, Ogden C, Fisher C, Corbishley C, De Bono J, Eeles R, Brewer D, Cooper C (2008) Detection of TMPRSS2-ERG translocations in human prostate cancer by expression profiling using GeneChip Human Exon 1.0 ST arrays. J Mol Diagn 10 (1): 50–57
King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL (2009) Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet 41 (5): 524–526
Koksal IT, Dirice E, Yasar D, Sanlioglu AD, Ciftcioglu A, Gulkesen KH, Ozes NO, Baykara M, Luleci G, Sanlioglu S (2004) The assessment of PTEN tumor suppressor gene in combination with Gleason scoring and serum PSA to evaluate progression of prostate carcinoma. Urol Oncol 22 (4): 307–312
Li Y, Cozzi PJ (2007) MUC1 is a promising therapeutic target for prostate cancer therapy. Curr Cancer Drug Targets 7 (3): 259–271
Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM, Shah RB (2007) Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol 20 (5): 538–544
Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, Shah RB, Gaston S, Tomlins SA, Wei JT, Kearney MC, Johnson LA, Tang JM, Chinnaiyan AM, Rubin MA, Sanda MG (2009) Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res 15 (14): 4706–4711
Nakagawa T, Kollmeyer TM, Morlan BW, Anderson SK, Bergstralh EJ, Davis BJ, Asmann YW, Klee GG, Ballman KV, Jenkins RB (2008) A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS ONE 3 (5): e2318
Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, Yang LY, Stanimirovic A, Encioiu E, Neill M, Loblaw DA, Trachtenberg J, Narod SA, Seth A (2007) Expression of the TMPRSS2: ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer 97 (12): 1690–1695
Narod SA, Seth A, Nam R (2008) Fusion in the ETS gene family and prostate cancer. Br J Cancer 99 (6): 847–851
Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD, Pienta KG, Kuefer R, Vessella R, Sun XW, Meyerson M, Lee C, Sellers WR, Chinnaiyan AM, Rubin MA (2006) TMPRSS2: ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res 66 (17): 8337–8341
Schmitz M, Grignard G, Margue C, Dippel W, Capesius C, Mossong J, Nathan M, Giacchi S, Scheiden R, Kieffer N (2007) Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int J Cancer 120 (6): 1284–1292
Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, Perner S, Sboner A, Pawitan Y, Andren O, Johnson LA, Tang J, Adami HO, Calza S, Chinnaiyan AM, Rhodes D, Tomlins S, Fall K, Mucci LA, Kantoff PW, Stampfer MJ, Andersson SO, Varenhorst E, Johansson JE, Brown M, Golub TR, Rubin MA (2008) Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst 100 (11): 815–825
Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, Shaheduzzaman S, Tan SH, Vaidyanathan G, Whitman E, Hawksworth DJ, Chen Y, Nau M, Patel V, Vahey M, Gutkind JS, Sreenath T, Petrovics G, Sesterhenn IA, McLeod DG, Srivastava S (2008) TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 27 (40): 5348–5353
Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM (2005) Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310 (5748): 644–648
Tu JJ, Rohan S, Kao J, Kitabayashi N, Mathew S, Chen YT (2007) Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod Pathol 20 (9): 921–928
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98 (9): 5116–5121
Wang J, Cai Y, Ren C, Ittmann M (2006) Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res 66 (17): 8347–8351
Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL (2009) Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 69 (3): 958–966
Yoshimoto M, Cutz JC, Nuin PA, Joshua AM, Bayani J, Evans AJ, Zielenska M, Squire JA (2006a) Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet Cytogenet 169 (2): 128–137
Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, Evans AJ, Zielenska M, Squire JA (2006b) Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia 8 (6): 465–469
Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O, Zielenska M, Soares FA, Squire JA (2008) Absence of TMPRSS2: ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol 21 (12): 1451–1460
Acknowledgements
We acknowledge the Emory Biomarker Service Center for its contribution to this research. This research is funded in part by the Canadian Cancer Society (Grant no. 019038).
Data deposition: Gene expression data is deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/) under series GSE18655.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website (http://www.nature.com/bjc)
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Barwick, B., Abramovitz, M., Kodani, M. et al. Prostate cancer genes associated with TMPRSS2–ERG gene fusion and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer 102, 570–576 (2010). https://doi.org/10.1038/sj.bjc.6605519
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605519
Keywords
This article is cited by
-
A harmonized resource of integrated prostate cancer clinical, -omic, and signature features
Scientific Data (2023)
-
Detecting survival-associated biomarkers from heterogeneous populations
Scientific Reports (2021)
-
Predictive significance of TMRPSS2-ERG fusion in prostate cancer: a meta-analysis
Cancer Cell International (2018)
-
Genetic association analysis of the RTK/ERK pathway with aggressive prostate cancer highlights the potential role of CCND2 in disease progression
Scientific Reports (2017)
-
The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer
Nature Communications (2014)
