
Abstract
MicroRNAs (miRNAs) are short non-coding RNAs that have key functions in a wide array of critical cell processes, including haematopoiesis by regulating the expression of multiple genes. Aberrant miRNA expression has been described in acute myeloid leukaemia suggesting a role in leukaemogenesis. In this review we summarise the current knowledge.
Similar content being viewed by others


Main
Acute myeloid leukaemia (AML) arises from myeloid progenitor cells that are arrested at early stages of differentiation. It is a cytogenetically heterogeneous disorder with acquired recurrent chromosomal alterations detected in about 55% of adult patients, such as translocations (i.e. t(15;17); t(8;21); t(9;11)), inversions (i.e. inv(16)), deletions (i.e. del(7q)), trisomies (i.e. +8) and monosomies (i.e. −5/−7) (reviewed by Mrózek et al, 2004). Importantly, AML patients could be stratified according to the detected cytogenetic abnormalities in high-, intermediate- and low-risk cytogenetic groups (Mrózek et al, 2004). In the remaining 45% of cases of cytogenetically normal AML (CN-AML), a number of novel molecular abnormalities have been described, such as the internal tandem duplication (ITD) in the juxtamembrane domain or mutation in the second tyrosine kinase domain (TKD) of FMS-like tyrosine kinase 3 (FLT3) gene, mutations in the nucleophosmin (NPM1) gene, CCAAT/enhancer binding protein-α (CEBPA) gene and in the Wilms’ tumour gene and partial tandem duplication (PTD) of the mixed-lineage leukaemia (MLL) gene (reviewed by Mrózek et al, 2007). In addition to mutations, overexpression of ERG (v-ets erythroblastosis virus E26 oncogene homologue) and BAALC (brain and acute leukaemia, cytoplasmic) genes has been found in CN-AML (Tanner et al, 2001; Baldus et al, 2004). Like cytogenetics, molecular abnormalities in CN-AML not only have improved the classification of this now heterogeneous group of AMLs, but also have prognostic implications. For example, NPM1 mutations predict favourable outcome in young CN-AML in the absence of FLT3-ITD (reviewed by Mrózek et al, 2007). The presence of CEBPA mutations identifies a group of patients with a better prognosis within the young high-risk CN-AML group (patients with FLT3-ITD and/or wild-type (wt) NPM1) (Marcucci et al, 2008b). Otherwise, CN-AML patients with FLT3-ITD have a significantly inferior outcome compared to patients with FLT3-wt (reviewed by Mrózek et al, 2007). The level of FLT3-ITD mutant allele was also correlated with outcome, whereas the prognosis impact of FLT3-TKD is controversial. MLL-PTD has been associated with shorter complete remission duration or worse event-free survival (EFS) without effect on overall survival (reviewed by Mrózek et al, 2007). Many of these prognostic studies are difficult to interpret because of the co-existence of one or two mutations which have to be taken in consideration because of their probable interactions and influence on prognosis. Additional limitations arise from the low number of patients and treatment differences (reviewed by Gaidzik and Döhner, 2008).
Despite great progress, AML biology remained poorly understood. Over the past years, complementary DNA (cDNA) microarrays have been used to interrogate whole gene expression in AML samples. Indeed, several groups have reported distinctive signatures associated with particular cytogenetic and molecular groups of AML (Bullinger et al, 2004; Ross et al, 2004; Valk et al, 2004; Radmacher et al, 2006). Using unsupervised analyses, we identified novel subgroups of AMLs based on gene expression profiling (GEP) obtained using Affymetrix microarrays. Moreover GEP allowed to further classify previously defined cytogenetic subgroups. For example, GEP uncovered substantial heterogeneity in core binding factor (CBF) AMLs suggesting alternative mutations or deregulated pathways involved in transformation (Bullinger et al, 2007). In some cases, the GEP signatures were able to predict outcome (Bullinger et al, 2004; Ross et al, 2004; Valk et al, 2004). More importantly, a cDNA microarray study identified a gene expression signature that separated CN-AML into two prognostically relevant subgroups (Bullinger et al, 2004). The prognostic value of this signature was validated in a different set of CN-AML patients using a different microarray platform (Radmacher et al, 2006). These studies confirmed the possible applicability of GEP for outcome prediction in CN-AML. Despite this progress, focusing on known genes will likely not suffice to uncover the molecular puzzle of AML. The integration of a whole genome approach including non-coding RNAs may lead to an improved understanding of AML biology.
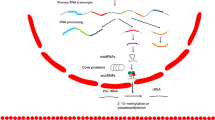
MicroRNAs (miRNAs) are non-coding RNAs of 19–25 nucleotides in length that regulate gene expression by repressing translation or accelerating mRNA decay (Bartel, 2004). MicroRNAs are involved in critical biological processes, including development, cell differentiation, apoptosis, proliferation and haematopoiesis (Chen et al, 2004; Poy et al, 2004; Xu et al, 2004; Cheng et al, 2005; Karp and Ambros, 2005). Recent data indicate that miRNAs are deregulated in diseases, such as diabetes, heart disease and cancer (Calin et al, 2002; Poy et al, 2004; van Rooij et al, 2006). The first study connecting miRNAs and leukaemia reported frequent down-regulation of the miRNA cluster; miR-15a/miR-16-1 in chronic lymphocytic leukaemia (CLL) (Calin et al, 2002). This cluster is localised at chromosome 13q14.3, a genomic region, which is deleted in about 65% of CLL patients. Further work showed that miR-15a/miR-16-1 cluster targets the anti-apoptotic BCL-2 (Cimmino et al, 2005). Thereby, this study provides with an alternative explanation for the BCL-2 up-regulation observed in CLL. Following earlier reports, two large miRNA profiling studies confirmed the widespread deregulation of miRNAs in cancer (Lu et al, 2005; Volinia et al, 2006). But how miRNAs contribute to oncogenesis? Further research established that miRNAs can behave as tumour suppressor genes or oncogenes (Calin and Croce, 2006; Garzon et al, 2006). An miRNA, which is down-regulated and targets an oncogene, acts as a tumour suppressor gene. For example, let-7a, which is down-regulated in lung adenocarcinoma, targets the highly expressed oncogene K-RAS (Johnson et al, 2005). In contrast, an miRNA which is over-expressed and targets a tumour suppressor gene acts as an oncogene (Calin and Croce, 2006; Garzon et al, 2006). For example, high expression of miR-155 and its host gene, the non-coding RNA B-cell integration cluster (BIC) has been reported in different human B-cell neoplasms suggesting a role for miR-155 in B-cell lymphomagenesis (Eis et al, 2005). This hypothesis was later confirmed by Costinean et al (2006) who reported an miR-155 transgenic mouse developing a polyclonal pre-leukaemic pre-B-cell proliferation evident in the spleen and bone marrow (BM) followed by a full-blown malignancy.
Over the past 3 years, several groups have described miRNA signatures associated with recurrent cytogenetic abnormalities, molecular aberrations and outcome in AML. In this review we discuss the current miRNA profiling studies in AML and outline future prospects and potential therapeutic applications.
miRNA expression in AML blasts, stem cells and committed progenitors
Four recent large-scale studies have reported miRNA expression in primary AML blasts and CD34+ selected cells (Garzon et al, 2008a; Jongen-Lavrencic et al, 2008) or BM samples (Dixon-McIver et al, 2008; Li et al, 2008) from healthy donors. Garzon et al (2008a) identified 26 down-regulated miRNAs and none up-regulated miRNAs in 122 newly diagnosed AML samples compared with BM CD34+ cells from 10 normal donors using a custom microarray platform. This is consistent with the miRNA profiling results obtained from 334 tumours, including leukaemias, and normal tissues using a bead-based flow cytometric miRNA platform (Lu et al, 2005). Lu et al (2005) found in general a lower expression of miRNAs in tumours compared with normal tissues. They hypothesised that global miRNA expression might reflect the state of cellular differentiation. Indeed, they observed in haematopoietic progenitor cells undergoing erythroid differentiation an increase in miRNA expression at a later stage of differentiation (Lu et al, 2005).
A second AML study analysed 215 newly diagnosed leukaemic patients and CD34+ cells from four healthy subjects using a multiplexing real-time quantitative polymerase chain reaction (qRT-PCR) method (Jongen-Lavrencic et al, 2008). Only 11 miRNAs (5 up- and 6 down-regulated) were found differentially expressed in normal CD34+ specimens with respect to the leukaemias. Among them, miR-21 was found up-regulated in AML samples compared with CD34+ cells.
The two other studies compared miRNA expression in AML samples with BM from healthy donors. Dixon-McIver et al (2008) showed the significant deregulation of 33 miRNAs (17 up-regulated and 16 down-regulated) in 100 leukaemia samples with respect to two BM samples using a qRT-PCR assay. Li et al (2008) using bead-based miRNA expression profiling assays analysed 54 AML samples and 3 normal BM samples. Normal control samples were grouped together in a subcluster with unsupervised analysis.
Only few miRNAs were commonly (in at least two studies) down-regulated in AML patients, such as miR-29b and miR-126. Many factors might be involved in the disparities of the results from these studies. First, the purity of the leukaemic cells might influence the results. There could be differences in the blast percentage among the studies that could impact on the profiling. In one study, selection purity was improved by CD3/CD19 depletion after Ficoll separation of mononuclear cells. Second, the nature of the control sample is critical and can also explain the discrepancies. Two studies used BM CD34+ cells whereas the other two used whole BM or mononuclear cells. Third, other factors such as biology differences in the AMLs included in the studies, number of control samples, processing, platform and bioinformatics approaches (data normalisation, filtering and clustering) may influence the results. Further studies will be needed using highly pure populations, including CD34+ selected leukaemic cells, to assess miRNA changes with respect to CD34+ normal cells.
miRNA signatures associated with cytogenetic and molecular alterations in AML
Garzon et al (2008a)analysed the miRNA expression in a cohort of 122 newly diagnosed primary AML samples with intermediate and poor prognosis using a custom miRNA platform. The authors identified miRNA expression profiles closely linked to 11q23 translocations, trisomy 8 and FLT3-ITD (Tables 1 and 2). Among the miRNAs down-regulated in balanced 11q23 translocation patients, many are tumour suppressor miRNAs that target critical oncogenes, i.e., miR-34b (CDK4 and CCNE2), miR-15a (BCL-2), the let-7 family (RAS), the miR-29 family (MCL-1 and TCL-1) and miR-372 (LATS2). Interestingly, miR-155 was found up-regulated in AML patients with high white count and FLT3-ITD mutations. This miRNA has been recently described to block ‘in vitro’ human myeloid colony formation, halt megakaryopoiesis (Romania et al, 2008) and induce a myeloproliferative disease in mice (O’Connell et al, 2008). A second study by Jongen-Lavrencic et al (2008) reported miRNA profiles using a qRT-PCR platform in 215 AML patients that include among others good risk karyotypes such as t(15;17), t(8;21) and inv(16). Using a combination of unsupervised and supervised analyses the authors identified distinctive miRNA expression profiles associated with known AML cytogenetic subtypes (Tables 1 and 2). Among them, the tumour suppressor miRNAs let-7b and let-7c (known to target the oncogenes RAS) were found down-regulated in CBF leukaemias (Johnson et al, 2005). None of these deregulated miRNAs was localised in the re-arranged chromosomal regions. Finally, the authors determined the minimal set of miRNAs and mRNAs (by performing GEP on the same AML cohort) that could predict a particular genetic subtype of AML. The mRNA-GEP class predictor was accurate in predicting AML with t(15;17), t(8;21) or inv(16) as well as CN-AML samples with NPM1, CEBPA mutations and FLT3-ITD. Another study using a qRT-PCR assay reported miRNA signatures that correlate with the most frequent cytogenetic alterations (t(15;17), t(8;21), inv(16), 11q23 translocations) in 100 AML samples. In particular, acute promyelocytic leukaemias bearing a t(15;17) had a distinctive signature including the up-regulation of seven miRNAs located on chromosome 14q32 which is not implicated in the translocation (Table 1; Dixon-McIver et al, 2008). More recently, the study of Li et al using another genome-wide bead-based miRNA expression platform analysed 52 AML samples with common translocations including t(8;21), inv(16), t(15;17) and MLL rearrangements (Table 1).
We reported in Table 1 the most significant miRNAs deregulated in association with the different cytogenetic subtypes in these four studies. In particular, we showed common miRNAs found up- or down-regulated by at least two studies in each subtypes (Table 1).
Li et al determined the minimal number of miRNAs that can accurately discriminate AML subtypes. Two (miR-126 and miR-126*), three (miR-224, miR-368 and miR-382) and seven miRNAs (from the polycistronic miR17-92 cluster) are sufficient to predict CBF AML, t(15;17) and MLL AML respectively, resulting in a diagnosis accuracy better than 94% (Table 1). Some of these differentially expressed miRNAs were also identified in the class predictor reported by Jongen-Lavrencic including miR-126* in CBF leukaemias or miR-382 in t(15;17) AML.
Garzon et al (2008b)focused on CN-AML with NPM1 mutation which is the most common genetic alteration in CN-AML. They profiled 85 de novo AML patients (55 with NPM1 mutation and 30 with NPM1 wt) and found a strong miRNA signature that distinguishes CN-AML with NPM1 mutation from NPM1 wt (Table 2). The up-regulation of miR-10a and miR-10b in NPM1-mutated AML clearly differentiate the two different forms. These results were confirmed by Jongen-Lavrencic et al and in a recent study of 189 older CN-AML (Becker et al, 2009). Among the other miRNAs up-regulated, three families of tumour suppressor miRNAs were involved: miR-15a/16-1, several let-7 and miR-29 members whereas miR-204 and miR-128a were down-regulated (Garzon et al, 2008b). Concerning CEBPA mutations, Marcucci et al (2008b) reported an miRNA signature associated with the presence of the CEBPA mutation in CN-AML patients. Interestingly, miR-181a and miR-181b were up-regulated in CEBPA-mutated cases. These results are consistent with a CEBPA mutation miRNA signature reported by Jongen-Lavrencic.
Prognostic role of mirnas in AML
Few studies have correlated miRNA expression changes with prognosis in AML. Garzon et al (2008a) first reported that in a relatively older AML cohort of patients (median age 59) with intermediate- and poor-risk cytogenetics, patients with high expression of miR-199a and miR-191 had a significant shorter overall survival and EFS. These results were validated in a second cohort of 60 AML patients with similar characteristics using a different technology; qRT-PCR. These two miRNAs (miR-191 and miR-199a) predicted outcome independent from other variables, including age and cytogenetics. Marcucci et al (2008a) recently showed an miRNA signature that correlated with EFS in young CN-AML patients (<60 years) with high-risk molecular features (defined by the detection of FLT3-ITD and/or an NPM1 wt). This subgroup of AML represents more or less a third of young AML patients. The prognostic signature included miR-181a and miR-181b which were inversely correlated with risk of event and miR-124, miR-128-1, miR-194, miR-219-5p, miR-220a, miR-320 which were positively associated with the risk of event. This prognostic signature was independent from other factors. Furthermore, using mRNA gene-chip microarray analysis, the authors showed an inverse correlation between the expression of miR-181 and predicted target genes. Many of these genes encode for proteins involved in pathways of innate immunity mediated by Toll-like receptors (TLR2, TLR4, TLR8) and interleukin-1β (NOD-like receptors CARD8, 12, 15 and CASP1). Activation of Toll-like receptors induces production of inflammatory cytokines through nuclear factor κB and interleukin-1β promotes the survival and the proliferation of AML blasts. No miRNA was found to be associated with outcome in low-risk group (NPM1 mutation alone) (Marcucci et al, 2008a). Both prognostic signatures need to be confirmed in large prospective studies before clinical applications. The different prognostic miRNA signatures between the two studies can be explained by two main factors. First, the median age between the two studies was different; 60.3 years (range 18–86) vs 45 years (range 19–45). This is critical because FLT3-ITD and NPM1 prognostic implications are age dependent. FLT3-ITD has been shown to have a negative prognostic impact only in young AML patients (reviewed by Mrózek et al, 2007), whereas it is not prognostic in the elderly (Stirewalt et al, 2001; Garzon et al, 2008a). NPM1 mutation seems to be an independent prognostic factor in young and older AML patients but only in the absence of FLT3-ITD in the young patients cohort (reviewed by Gaidzik and Döhner, 2008; Becker et al, 2009). Second, the cytogenetic/molecular subgroups studied were different. Garzon et al (2008a) studied intermediate- and poor-risk cytogenetic groups, whereas Marcucci et al (2008a) focused on high-risk CN-AML.
miRNAs and HOX genes
HOX genes are transcription factors which are important in the regulation of early stages of haematopoiesis, including self-renewal of haematopoietic stem cells (Sauvageau et al, 1995). Five miRNAs are embedded in the HOX genes cluster – miR-196b in HOXA (at chromosome 7p15), miR-10a and 196a-1 in HOXB (at chromosome 17q21), miR-196a-2 in HOXC (at 12q13) and miR-10b in HOXD (at 2q31) clusters. A distinctive correlation between the miR-10a, miR-10b and miR-196-1 and the HOXA/HOXB gene expression was first reported in 30 AML patients suggesting a common regulatory mechanism (Debernardi et al, 2007). Kuchenbauer et al (2008) recently showed an up-regulation of all the miRNAs located in the HOX cluster in a murine leukaemia model (engineered by over-expression of NUP98/HOXD13 fusion gene with the oncogenic collaborator MEIS1).
Npm1, mirnas and hox genes
Interestingly, miR-10a, 10b and miR-196b have recently been linked to molecular or chromosomal alterations in subtypes of AML, in particular CN-AML with mutated NPM1. Garzon et al (2008b) identified a statistically significant up-regulation miR-10a, miR-10b in CN-AML with NPM1 mutation. The over-expression was correlated with high expression level of HOX genes (mainly HOXB genes) (Garzon et al, 2008b). These results were validated by other groups (Debernardi et al, 2007; Jongen-Lavrencic et al, 2008; Becker et al, 2009). Whether these miRNAs are merely bystanders or involved in leukaemogenesis is still under investigation. Among the miRNAs down-regulated in NPM1-mutated AML, miR-204 directly targets HOXA10 and MEIS1 (Garzon et al, 2008b). Over-expression of HOXA10 in murine haematopoietic stem cells was reported to perturb myeloid differentiation and to lead to AML (Thorsteinsdottir et al, 1997). Similar outcome was shown by over-expressing HOXA9 and MEIS1 in mice (Kroon et al, 1998). The data suggest that miRNAs may be responsible in part of the HOX up-regulation observed in NPM1-mutated AML.
MLL- and HOX-embedded miRNAs
Popovic et al (2009) reported that miR-196b is over-expressed specifically in AML with MLL rearrangement. The MLL gene (located in 11q23) is commonly involved in chromosomal translocations (with more than 60 different fusion partners reported) responsible of AML. Leukaemias related with MLL are in most of the cases characterised by an over-expression of a subset of HOX genes including HOXA9 (Rozovskaia et al, 2001). MLL binds to specific clusters of CpG islands of HOXA9, adjacent to miR-196b, and maintains its expression by protecting these clusters from DNA methylation (Erfurth et al, 2008). Recently, Popovic et al (2009) showed that MLL regulates miR-196b expression similarly to the surrounding HOX genes. Moreover, MLL fusion proteins cause up-regulation of miR-196b in primary BM cells (Popovic et al, 2009). MiR-196b over-expression seems to increase proliferation, survival and partially block the differentiation of normal BM haematopoietic progenitor cells suggesting importance of miR-196b in MLL leukaemias. It is noteworthy that the MLL fusion proteins have also been shown to interact directly with Drosha, the nuclear RNase III enzyme crucial in miRNA biogenesis (Nakamura et al, 2007).
miRNAs and epigenetics
Recent data indicate that miRNAs are targets of aberrant epigenetics in AML. The ETO/AML1 is the fusion protein resulting from the t(8;21) translocation detected in about 15% of AML. The in vitro expression of ETO/AML1 in haematopoietic stem cells leads to the expansion of the myeloid progenitors and a pre-leukaemic state. Fazi et al (2007) showed that the binding of AML1 to the pre-miR-223 promoter is responsible for miR-223 transcription activation. The ETO/AML1 protein can bind to this site and activate histone deacetylates and DNA methyltransferases, thereby blocking the transcription of miR-223 by inducing histone deacetylation and increasing DNA methylation.
Our group recently reported that over-expressing miR-29b in AML cell lines and primary AML blasts down-regulates the expression of DNA methyltransferases; DNMT1, DNMT3A and 3B. MiR-29b targets directly DNMT3A and 3B and indirectly DNMT1 through its activator Sp1. Restoration of miR-29b leads to global DNA hypomethylation and re-expression of tumour suppressor genes such as the cyclin-dependent kinase inhibitor p15INK4b and the oestrogen receptor ESR1 genes (Garzon et al, 2009).
Discussion and future directions
In this review, we described four recent large-scale global miRNA profiling studies in AML. The differences among the miRNA signatures reported by these studies can be explained mainly by the use of various cytogenetic and molecular AML subgroups and by the type of platform used to interrogate miRNA expressions. The frequencies of cytogenetic/molecular abnormalities were clearly different among the studies. For example, Garzon et al focused on intermediate and poor prognostic cytogenetic groups of AML, whereas Jongen-Lavrencic included good risk subgroups such as t(8;21), inv(16) and t(15;17). Because most of the strategies to identify distinctive miRNA signatures were based on two-way comparisons (i.e. inv(16) vs all other AMLs), the heterogeneity of the other AMLs will impact greatly on the analysis. This bias is less evident when the authors focus on well-characterised cytogenetic and molecular subgroups of AML. Common miRNAs have been found deregulated in CN-AML cases with FLT3-ITD, NPM1 and CEBPA mutated and in patients with t(15;17), inv(16) and t(8;21). These results are significant because they will provide with a set of validated miRNAs whose functions will be further investigated. These studies may lead to a better molecular classification, understanding of AML biology and discovery of novel targets for treatment. In addition to improving diagnosis, miRNA signatures may enhance current prognostic markers and help to better stratify patients for treatment. But before any clinical applications, miRNA profiling studies require to be extensively validated and more standard, reliable and reproducible platforms will need to be developed. Future miRNA expression studies should use very well cytogenetically and molecularly characterised AML subtypes to minimise biology heterogeneity. Generation of small RNA libraries and next-generation deep sequencing will enable not only to quantify the whole miRNA transcripts but also to obtain sequences information and discover new miRNAs.
We also believe that miRNAs studies have potential relevant therapeutic implications. Synthetic miRNAs or anti-miRNAs alone or in association with chemotherapy could be promising in future AML therapies. Synthetic miRNAs can be used to restore lost tumour suppressor miRNAs expression. The other strategy (so far more developed) is to silence oncogenic miRNAs (with ‘antagomirs’ or anti-miRNA oligonucleotides or locked nucleic-acid-modified oligonucleotides) which are over-expressed in AML (Krützfeldt et al, 2005). Moreover, the impact of some drugs, such as DNA-demethylating agents, on miRNAs expression is also another therapeutic approach to modulate miRNA expression. If miRNA-based therapy offers new perspectives, several issues have still to be considered such as off-target effects, chemistry considerations and target delivery. A better comprehension of the regulation and function of the miRNAs will be necessary to develop future miRNA-based new drugs.
Conclusion
Altogether, the data suggest that miRNAs may complement and enhance our current knowledge about AML. MicroRNA expression profiling has been associated with cytogenetic and molecular subtypes of AML. Functional studies suggested miRNAs may play a role in AML pathogenesis being involved in essential pathways for myeloid differentiation. It is now well demonstrated that miRNAs represent a class of genes with a great potential for diagnosis, prognosis and therapy in AML. MicroRNA-based therapy will certainly play a role in the future of treatment of AML.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baldus CD, Liyanarachchi S, Mrózek K, Auer H, Tanner SM, Guimond M, Ruppert AS, Mohamed N, Davuluri RV, Caligiuri MA, Bloomfield CD, de la Chapelle A (2004) Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: amplification discloses overexpression of APP, ETS2, and ERG genes. Proc Natl Acad Sci USA 101 (11): 3915–3920
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 (2): 281–297
Becker H, Marcucci G, Maharry K, Margeson D, Radmacher MD, Whitman SP, Mrózek K, Baer MR, Larson RA, Bloomfield CD for Cancer and Leukemia Group B (CALGB) (2009) NPM1 mutations as an independent prognosticator for older cytogenetically normal acute myeloid leukemia (CN AML). J Clin Oncol 27(15S): abstract 7000
Bullinger L, Döhner K, Bair E, Fröhling S, Schlenk RF, Tibshirani R, Döhner H, Pollack JR (2004) Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med 350 (16): 1605–1616
Bullinger L, Rücker FG, Kurz S, Du J, Scholl C, Sander S, Corbacioglu A, Lottaz C, Krauter J, Fröhling S, Ganser A, Schlenk RF, Döhner K, Pollack JR, Döhner H (2007) Gene-expression profiling identifies distinct subclasses of core binding factor acute myeloid leukemia. Blood 110 (4): 1291–1300
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM (2002) Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 99 (24): 15524–15529
Calin GA, Croce CM (2006) MicroRNA-cancer connection: the beginning of a new tale. Cancer Res 66 (15): 7390–7394
Chen CZ, Li L, Lodish HF, Bartel DP (2004) MicroRNAs modulate hematopoietic lineage differentiation. Science 303 (5654): 83–86
Cheng AM, Byrom MW, Shelton J, Ford LP (2005) Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33 (4): 1290–1297
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102 (39): 13944–13949
Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM (2006) Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci USA 103 (18): 7024–7029
Debernardi S, Skoulakis S, Molloy G, Chaplin T, Dixon-McIver A, Young BD (2007) MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 21 (5): 912–916
Dixon-McIver A, East P, Mein CA, Cazier JB, Molloy G, Chaplin T, Andrew Lister T, Young BD, Debernardi S (2008) Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS ONE 3 (5): e2141
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE (2005) Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA 102 (10): 3627–3632
Erfurth FE, Popovic R, Grembecka J, Cierpicki T, Theisler C, Xia ZB, Stuart T, Diaz MO, Bushweller JH, Zeleznik-Le NJ (2008) MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc Natl Acad Sci USA 105 (21): 7517–7522
Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L, Diverio D, Ammatuna E, Cimino G, Lo-Coco F, Grignani F, Nervi C (2007) Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 12 (5): 457–466
Gaidzik V, Döhner K (2008) Prognostic implications of gene mutations in acute myeloid leukemia with normal cytogenetics. Semin Oncol 35 (4): 346–355
Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM (2006) MicroRNA expression and function in cancer. Trends Mol Med 12 (12): 580–587
Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K, Alder H, Nakamura T, Flomenberg N, Marcucci G, Calin GA, Kornblau SM, Kantarjian H, Bloomfield CD, Andreeff M, Croce CM (2008a) MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 111 (6): 3183–3189
Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, Volinia S, Liu CG, Schnittger S, Haferlach T, Liso A, Diverio D, Mancini M, Meloni G, Foa R, Martelli MF, Mecucci C, Croce CM, Falini B (2008b) Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA 105 (10): 3945–3950
Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu LC, Croce CM, Marcucci G (2009) MicroRNA -29b induces global DNA hypomethylation and tumor suppressor gene re-expression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113 (25): 6411–6418
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ (2005) RAS is regulated by the let-7 microRNA family. Cell 120 (5): 635–647
Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Löwenberg B (2008) MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 111 (10): 5078–5085
Karp X, Ambros V (2005) Developmental biology. Encountering microRNAs in cell fate signaling. Science 310 (5752): 1288–1289
Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G (1998) Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 17 (13): 3714–3725
Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438 (7068): 685–689
Kuchenbauer F, Morin RD, Argiropoulos B, Petriv OI, Griffith M, Heuser M, Yung E, Piper J, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, Hansen CL, Marra MA, Humphries RK (2008) In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res 18 (11): 1787–1797
Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, Neilly MB, Jin J, Zhang Y, Bohlander SK, Zhang DE, Larson RA, Le Beau MM, Thirman MJ, Golub TR, Rowley JD, Chen J (2008) Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA 105 (40): 15535–15540
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435 (7043): 834–838
Marcucci G, Radmacher MD, Maharry K, Mrózek K, Ruppert AS, Paschka P, Vukosavljevic T, Whitman SP, Baldus CD, Langer C, Liu CG, Carroll AJ, Powell BL, Garzon R, Croce CM, Kolitz JE, Caligiuri MA, Larson RA, Bloomfield CD (2008a) MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1919–1928
Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P, Whitman SP, Langer C, Baldus CD, Liu CG, Ruppert AS, Powell BL, Carroll AJ, Caligiuri MA, Kolitz JE, Larson RA, Bloomfield CD (2008b) Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078–5087
Mrózek K, Heerema NA, Bloomfield CD (2004) Cytogenetics in acute leukemia. Blood Rev 18 (2): 115–136
Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD (2007) Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 109 (2): 431–448
Nakamura T, Canaani E, Croce CM (2007) Oncogenic All1 fusion proteins target Drosha-mediated microRNA processing. Proc Natl Acad Sci USA 104 (26): 10980–10985
O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D (2008) Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 205 (3): 585–594
Popovic R, Riesbeck LE, Velu CS, Chaubey A, Zhang J, Achille NJ, Erfurth FE, Eaton K, Lu J, Grimes HL, Chen J, Rowley JD, Zeleznik-Le NJ (2009) Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 113 (14): 3314–3322
Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432 (7014): 226–230
Radmacher MD, Marcucci G, Ruppert AS, Mrózek K, Whitman SP, Vardiman JW, Paschka P, Vukosavljevic T, Baldus CD, Kolitz JE, Caligiuri MA, Larson RA, Bloomfield CD, Cancer and Leukemia Group B (2006) Independent confirmation of a prognostic gene-expression signature in adult acute myeloid leukemia with a normal karyotype: a Cancer and Leukemia Group B Study. Blood 108 (5): 1677–1683
Romania P, Lulli V, Pelosi E, Biffoni M, Peschle C, Marziali G (2008) MicroRNA 155 modulates megakaryopoiesis at progenitor and precursor level by targeting Ets-1 and Meis1 transcription factors. Br J Haematol 143 (4): 570–580
Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, Ribeiro RC, Rubnitz JE, Girtman K, Williams WK, Raimondi SC, Liang DC, Shih LY, Pui CH, Downing JR (2004) Gene expression profiling of pediatric acute myelogenous leukemia. Blood 104 (12): 3679–3687
Rozovskaia T, Feinstein E, Mor O, Foa R, Blechman J, Nakamura T, Croce CM, Cimino G, Canaani E (2001) Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4;11) abnormality. Oncogene 20 (7): 874–878
Sauvageau G, Thorsteinsdottir U, Eaves CJ, Lawrence HJ, Largman C, Lansdorp PM, Humphries RK (1995) Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev 9 (14): 1753–1765
Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, Radich JP (2001) FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood 97 (11): 3589–3595
Tanner SM, Austin JL, Leone G, Rush LJ, Plass C, Heinonen K, Mrózek K, Sill H, Knuutila S, Kolitz JE, Archer KJ, Caligiuri MA, Bloomfield CD, de La Chapelle A (2001) BAALC, the human member of a novel mammalian neuroectoderm gene lineage, is implicated in hematopoiesis and acute leukemia. Proc Natl Acad Sci USA 98 (24): 13901–13906
Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, Largman C, Humphries RK (1997) Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol Cell Biol 17 (1): 495–505
Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Löwenberg B, Delwel R (2004) Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 350 (16): 1617–1628
van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN (2006) A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103 (48): 18255–18260
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 103 (7): 2257–2261
Xu P, Guo M, Hay BA (2004) MicroRNAs and the regulation of cell death. Trends Genet 20 (12): 617–624
Acknowledgements
VH was supported by a grant Télévie from the Belgian Fonds National de la Recherche Scientifique and RG was supported by the Kimmel Foundation for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Havelange, V., Garzon, R. & Croce, C. MicroRNAs: new players in acute myeloid leukaemia. Br J Cancer 101, 743–748 (2009). https://doi.org/10.1038/sj.bjc.6605232
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605232
Keywords
This article is cited by
-
A novel computational method enables RNA editome profiling during human hematopoiesis from scRNA-seq data
Scientific Reports (2023)
-
miR-603 promotes cell proliferation and differentiation by targeting TrkB in acute promyelocytic leukemia
Annals of Hematology (2023)
-
Leukemic stem cell signatures in Acute myeloid leukemia- targeting the Guardians with novel approaches
Stem Cell Reviews and Reports (2022)
-
The crucial choice of reference genes: identification of miR-191-5p for normalization of miRNAs expression in bone marrow mesenchymal stromal cell and HS27a/HS5 cell lines
Scientific Reports (2020)
-
Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia
Leukemia (2015)
