
Abstract
Background:
Insulin-like growth factor (IGF)-I signalling stimulates proliferation, survival, and invasion in malignant mesothelioma and other tumour types. Studies have found that tumourigenesis is linked to dysregulation of cap-dependent protein translation.
Methods:
The effect of IGF stimulation on cap-mediated translation activation in mesothelioma cell lines was studied using binding assays to a synthetic 7-methyl GTP-cap analogue. In addition, cap-mediated translation was genetically repressed in these cells with a dominant active motive of 4E-BP1.
Results:
In most mesothelioma cell lines, IGF-I stimulation resulted in a hyperphosphorylation-mediated inactivation of 4E-BP1 compared with that in normal mesothelial cells. An inhibitor of Akt diminished IGF-I-mediated phosphorylation of 4E-BP1, whereas inhibiting MAPK signalling had no such effect. IGF-I stimulation resulted in the activation of the cap-mediated translation complex as indicated by an increased eIF4G/eIF4E ratio in cap-affinity assays. Akt inhibition reversed the eIF4G/eIF4E ratio. Mesothelioma cells transfected with an activated 4E-BP1 protein (4E-BP1A37/A46) were resistant to IGF-I-mediated growth, motility, and colony formation. In a murine xenograft model, mesothelioma cells expressing the dominant active 4E-BP1A37/A46 repressor protein showed abrogated tumourigenicity compared with control tumours.
Conclusion:
IGF-I signalling in mesothelioma cells drives cell proliferation, motility, and tumourigenesis through its ability to activate cap-mediated protein translation complex through PI3K/Akt/mTOR signalling.
Similar content being viewed by others


Main
Malignant mesothelioma is an aggressive cancer arising from the serosal lining of the pleural or peritoneal cavity. Along with asbestos exposure, simian virus 40 infection has been implicated in the development of mesothelioma. Despite attempts to treat mesothelioma with multimodal therapies, estimates of median survival are usually less than 1 year (Whitson and Kratzke, 2006; Lee et al, 2007). In the United States, nearly 3000 deaths are attributed to mesothelioma annually (Britton, 2002). The pathogenesis of mesothelioma involves multiple-signalling pathways, including vascular endothelial growth factor, epidermal growth factor, wnt, and insulin-like growth factor-I (IGF-I) (Whitson and Kratzke, 2006; Lee et al, 2007). A recent study has also demonstrated that both the Ras/mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3′ kinase (PI3K)/Akt pathway are activated in mesothelioma cells compared with non-transformed mesothelial cells (Patel et al, 2007). Many studies have found that the risk of developing several common cancers is linked to elevated levels of circulating IGF-I, as well as to an increased ratio of IGF-I to inhibitory-binding proteins (reviewed in (Jerome et al, 2003)). An array-based study found the presence of altered transcript profiles of genes from the IGF-I axis (Hoang et al, 2004a). In a follow-up study, mRNA levels of 12 components of the IGF-I axis were elevated in mesothelioma cells compared with that in a mesothelial cell line (Hoang et al, 2004b). Four mRNA levels, in particular, were increased: IGF-I (43.3-fold), insulin receptor substrate (IRS)-2 (24.0-fold), IRS-1 (4.4-fold), and IGF1R (2.6-fold). This suggests that an activated IGF-I axis contributes to the malignant phenotype of mesothelioma. In addition, the activation of IGF1R adaptor proteins (IRS-1 and IRS-2) has been shown to contribute to the proliferative or motility phenotype of mesothelioma cells. These previous studies along with others (Lee et al, 1993) demonstrate the importance of the IGF-I system in the pathogenesis of mesothelioma.
In normal cells, IGF-I binds to IGF1R and activates its kinase activity, leading to the targeting of its corresponding adaptor proteins, including IRS-1, IRS-2, and SHC. These substrates initiate phosphorylation cascades, resulting in the activation of the extracellular signal-regulated kinase (ERK) 1/2 and PI3K/Akt pathways and leading to cell proliferation and survival (reviewed in (Sachdev and Yee, 2006, Haddad and Yee, 2006)). Among the probable downstream targets of these pathways is the cap-mediated translation complex. In IGF-I-driven cancers, it is highly probable that the cancer-specific effects of IGF1R activation are mediated in part through the activation of cap-mediated translation.
Deranged cap-dependent protein translation is implicated in tumourigenesis in multiple tumour types (Jacobson et al, 2006), including mesothelioma (Patel et al, 2007). The rate-limiting step in the initiation of mRNA translation is the binding of eukaryotic initiation factor 4E (eIF4E) to the 5′-cap structure (m7GpppN) of mRNA. Overexpression of eIF4E is observed in many solid tumours. Under normal cellular conditions, eIF4E is negatively regulated by 4E-BP proteins. 4E-BP1 blocks the interaction between eIF4E and the scaffolding protein eIF4G, inhibiting the formation of the active eIF4F complex and suppressing cap-mediated translation. 4E-BP1 undergoes hierarchical phosphorylation during mitogenic stimulation, which decreases its affinity for eIF4E and results in the activation of translation (De Benedetti and Graff, 2004; Polunovsky and Bitterman, 2006). In cancer, elevated levels of active eIF4F increase the translation of “weak” mRNAs with long structured 5′-untranslated regions, which typically encode anti-apoptotic proteins and growth regulatory factors contributing to tumourigenesis (Graff and Zimmer, 2003; Thumma and Kratzke, 2007).
This study examines the importance of cap-dependent translation in mediating the effects of the IGF-I axis in mesothelioma. Evidence is presented establishing the fact that activation of both PI3K and ERK1/2 pathways after IGF-I stimulation leads to a stimulation of the eIF4F complex. Furthermore, a dominant active 4E-BP1 results in the repression of translation and disrupts IGF-I-mediated motility and proliferation of mesothelioma cells. Expression of the dominant active 4E-BP1 resulted in significantly decreased tumourigenicity of murine xenografts. These results demonstrate that the malignant phenotype and tumourigenicity conferred upon mesothelioma by the IGF-I pathway is dependent upon the activation of cap-dependent translation.
Materials and methods
Cell lines and cell culture
Mesothelioma cell lines were obtained from either the American Type Culture Collection or from Frederick Kaye (NCI). The medium used for H513, H2052, H2373, H2461, and H2596 was RPMI (Gibco, Invitrogen, Carlsbad, CA, USA), containing 10% calf serum (Biofluids, Rockville, MD, USA). LP9 cells, a non-transformed human mesothelial cell line, were obtained from the National Institute on Aging Cell Repository at passage five. They were maintained in a medium containing a 1 : 1 ratio of M199 and MCDB10 basal medium (Sigma), supplemented with 15% calf serum, 10 ng ml−1 EGF, and 0.4 μg ml−1 hydrocortisone.
IGF-I stimulation was carried out on cells grown to 50% confluence. Cells were washed twice with phosphate-buffered saline (PBS) and grown in a serum-free medium (SFM) for 14–16 h. Also added at this time to the appropriate plates was the PI3K inhibitor, LY294002 (Promega, Madison, WI, USA) (20 μ M), or the MAPK kinase (MAPKK or MEK) inhibitor, U0126 (Promega) (10 μ M). The medium was then replaced with fresh SFM containing or not containing 5 nM IGF-I (Gro Pep) for the indicated times and, where indicated, fresh pathway inhibitors were also added. All cells were maintained at 37°C in 5% CO2.
Cell lysate preparation
Cell lysate was prepared for the cap-affinity assays as follows: Cells were washed with PBS, scraped from the plate in the presence of 1 ml PBS, centrifuged, and resuspended in five times the pellet volume of freeze–thaw lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 10 mM tetrasodium pyrophosphate), supplemented with a protease inhibitor cocktail (Roche, Basel, Switzerland) and a phosphatase inhibitor cocktail (Sigma). The cells were subjected to three consecutive freeze (15 min at −80°C) and thaw (2 min at 37°C) cycles, followed by centrifugation (14K r.p.m., 4°C) for 10 min. For the purpose of studying membrane-bound proteins, a second batch of cell lysate was prepared from cells processed under identical conditions but in the detergent-containing lysis buffer TNESV (50 mM Tris-HCl, pH 7.4; 1% NP-40; 2 mM EDTA, pH 8.0; 0.1 M NaCl), supplemented with both protease and phosphatase inhibitors. Lysate protein concentrations were determined by Bradford assay and stored at −80°C.
Immunoblot analysis
Protein samples were separated by SDS-PAGE on either straight 15% or an 8–15% gradient (cap-affinity assay samples) and then transferred to a PVDF membrane (Hybond-P, Amersham Biosciences, Piscataway, NJ, USA). Membranes were blocked in 5% non-fat dry milk for 1 h at room temperature in Tris-buffered saline Tween 20 (TBST: 0.15 M NaCl; 0.01 M Tris-HCl, pH 7.6; 0.05% Tween 20), rinsed, and incubated for 1 h at room temperature with the appropriate primary antibody. Blots were probed separately with either rabbit α-eIF4GI antibody (kindly provided by Nahum Sonenberg, McGill University Montreal, Quebec, Canada) at a 1 : 2500 dilution, rabbit α-IGF1R antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at a 1 : 1000 dilution, rabbit α-phospho-IGF1R antibody (Cell Signaling, Danvers, MA, USA) at a 1 : 1000 dilution, rabbit α-Akt antibody (Cell Signaling) at a 1 : 1000 dilution, rabbit α-phospho-Akt (ser473) antibody (Cell Signaling) at a 1 : 1000 dilution, rabbit α-MAPK antibody (Cell Signaling) at a 1 : 1000 dilution, rabbit α-phospho-MAPK (Thr202/Tyr204) antibody (Cell Signaling) at a 1 : 1000 dilution, rabbit α-4E-BP1 antibody (Abcam Inc., Cambridge, MA, USA) at a 1 : 2500 dilution, α-phospho-4E-BP1 (ser65) antibody (Cell Signaling) at a 1 : 1000 dilution, α-eIF4E antibody (BD Biosciences, San Jose, CA, USA) at a 1 : 500 dilution, mouse α-actin (Sigma) at a 1 : 10 000 dilution, or rat α-HA antibody (Roche) at a 1 : 2000 dilution to detect the haemagglutinin-tagged HA-4E-BP1A37/A46 proteins. The blots were washed thrice in TBST before incubation with appropriate horseradish peroxidase-labeled secondary antibodies, followed by incubation for 1 h at room temperature and three more washes in TBST. Bands of interest were visualised with the ECL Plus western blotting system (Amersham Biosciences, Piscataway, NJ, USA). The density of protein bands was determined using ImageJ, a public domain Java image-processing programme.
Cell transfection
Mesothelioma cell lines were transfected with the plasmid pACTAG-2 encoding the neomycin resistance gene or with pACTAG neo/HA-4E-BP1A37/A46. The cells were plated at 50% confluence and transfected (Lipofectin reagent (Invitrogen Life Technologies, Carlsbad, CA, USA)) with 5 μg of the appropriate plasmid DNA. After 72 h, the growth medium was replaced with medium containing G-418 (Cellgro, Manassas, VA, USA) (250 μg ml−1). Resistant colonies were collected and transferred to a fresh plate and propagated.
Cloning efficiency assay
Cell lines were transfected according to the procedure as described above, with one exception. When resistant colonies were of appropriate size, plates were fixed in 10% methanol and 5% acetic acid for 10 min, stained in 0.1% crystal violet/20% ethanol for 5 min, and scored for G-418-resistant colonies as previously described (Kratzke et al, 1995). All transfections were carried out in triplicate.
Cell migration assay
Cell motility was assessed by seeding 5 × 103 cells into coated wells of BioCoat Invasion Chambers (BD Biosciences). Twenty-four hours later, the cells were washed twice with PBS and cultured in SFM with or without 5 nM IGF-I. After 24 h of incubation, the cells remaining on the top of the membrane were removed with cotton swabs, whereas cells migrating through the 8 μm-sized pores onto the opposite side of the membrane were fixed (100% methanol), stained (0.1% crystal violet), and then counted microscopically. All experiments were carried out in triplicate.
Cell proliferation assay
Cells were seeded as triplicate sets into 96-well plates with 6000 cells per well for H2373, H2373 empty vector, and H2373 4E-BP1A37/A46, and with 3000 cells per well for H2461, H2461 empty vector, and H2461 4E-BP1A37/A46. After 24 h, the cells were switched to SFM and incubated for another 24 h in the presence or absence of 5 nM IGF-I. After incubation, cell growth was measured using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer's protocol. At the appropriate time, 10 μl of reagent was added to each well and incubated at 37°C for 4 h. Absorbance at 450 nm for each well was read on a microplate reader. Each experiment was repeated twice with similar results for each cell line.
In vitro cap-affinity assay
Cap-affinity assay was performed as before (Jacobson et al, 2006); briefly, 300 μl (1 μg μl−1) of cell lysate was incubated while mixing for 2 h at 4 °C with 50 μl of suspended (50% mixture) 7-methyl GTP-Sepharose4B (Amersham Biosciences) to capture eIF4E and its binding partners, eIF4G and 4E-BP1. The captured proteins were eluted with 35 μl of elution buffer (25 mM Tris-HCl pH 7.5, 150 mM KCl) containing 100 μ M of 7-methylguanosine 5′-triphosphate (Sigma-Aldrich, St Louis, MO, USA) and were prepared for immunoblotting.
Xenograft tumourigenicity assay
H2373 cells (2.1 × 107) stably transfected with pACTAG neo or with pACTAG neo/HA-4E-BP1A37/A46 were suspended in 0.5 ml of PBS and injected subcutaneously into the left or right flank, respectively, of five nude mice (nu/nu; Harlan). Tumour formation was quantified after 23 days by weight. Another set of five nude mice were injected with 0.5 ml of PBS containing H2596 cells (2.6 × 107) stably transfected with pACTAG neo or pACTAG neo/HA-4E-BP1A37/A46 into the left or right flank, respectively. Fourteen days after injection, the growth of these tumours was quantified by weight. All animal experiments were carried out under Institutional Animal Care and Use Committee of the Department of Veteran Affairs Medical Center, which conforms with the UKCCCR guidelines (Workman et al, 1998).
Results
IGF-I-mediated inactivation of 4E-BP1 depends on Akt signalling in mesothelioma
To assess the impact of IGF-I stimulation on the activation of signalling pathways underlying the malignant phenotype, a panel of mesothelioma cell lines and normal mesothelial cells were treated with IGF-I (5 nM) for varying times, followed by immunoblot analysis. Treatment of IGF-I for 20 min stimulated the intrinsic tyrosine kinase activity of IGF1R, resulting in phosphorylation in both mesothelioma cells and control LP9 mesothelial cells (Figure 1A). Increased IGF1R phosphorylation lasted for up to 300 min in two cell lines (H2461, H513), but drifted back to baseline levels in the remaining mesothelioma cells (H2373, H2052, and H2596) and control LP9 mesothelial cells. As expected, total IGF1R levels did not change during the experiment. As shown in earlier studies (Hoang et al, 2004b), MAPK and PI3K/Akt were phosphorylated upon IGF-I stimulation for 20 min in all five of the mesothelioma cell lines and control mesothelial cells. Peak levels of MAPK and Akt phosphorylation were reached at 20 min in all mesothelioma cell lines, with the exception of H2596 cells, which peaked at 60 min. Normal LP9 mesothelial cells had a weaker response to IGF-I stimulation. Total Akt and MAPK protein levels remained stable for both the LP9 control cells and mesothelioma cell lines.

IGF-I stimulation activates downstream-signalling pathways and inactivates 4E-BP1 in mesothelioma cell lines. (A) Immunoblot analysis showing phosphorylation and total levels of proteins involved in cell-signalling pathways (IGF1R, Akt, and MAPK) or in initiation of translation (eIF4G, eIF4E, and 4E-BP1) after treatment (in minutes) with and without IGF-I (5 nM) or on treatment with IGF-I (5 nM for 20 min) combined with LY249002 (LY) or U0126 (U). (B) Representation of the percentage of 4E-BP1 in hypophosphorylated isoforms (α, open columns) compared with that in hyperphosphorylated isoforms (β + γ, filled columns) for mesothelioma cells and mesothelial control cells (LP9) not treated or treated with IGF-I (5 nM) or IGF-I combined with LY249002 (LY) or U0126 (U) for the indicated times. β-actin represents loading controls for each cell line.
Sequential phosphorylation of 4E-BP1 leads to slower electrophoretic migration, with the γ, β, and α isoforms corresponding, respectively, to hyper-, intermediate, and hypophosphorylation. After IGF-I stimulation, slow-migrating γ and β isoforms of 4E-BP1 were phosphorylated in a sustained manner even for up to 300 min in most, but not all, of the mesothelioma cell lines (Figure 1A). The percentage of hypophosphorylated 4E-BP1 decreased when mesothelioma cells were stimulated with IGF-I (Figure 1B). These results reveal a direct correlation between IGF-I stimulation and inactivation of the 4E-BP1 repressor protein.
The roles of Akt and MAPK pathways were investigated next to delineate whether these pathways mediate an IGF-I-induced inactivation of 4E-BP1 in mesothelioma. Akt inhibition with LY294002 strongly diminished the phosphorylation of Akt and its downstream target 4E-BP1 in all five mesothelioma cell lines and control LP9 cells (Figure 1A and B). Although MAPK inhibition with U0126 did suppress the phosphorylation of MAPK, it did not suppress 4E-BP1 phosphorylation as expected (Figure 1A and B). These results confirm that IGF-I-mediated inactivation of 4E-BP1 is dependent upon Akt/mTOR signalling. Finally, the level of eIF4G, the scaffolding component of the eIF4F complex necessary for the initiation of translation, was not affected by IGF-I stimulation and was mildly decreased upon LY294002 treatment in three of the five mesothelioma cell lines (H2052, H513, and H2596) and LP9 cells (Figure 1A). It is known that eIF4G is cleaved by caspase 3 during apoptosis induction. It is possible that treatment with LY249002 decreased eIF4G levels by inducing caspase-mediated cleavage (Prevot et al, 2003).
IGF-I stimulation activates the cap-mediated translation complex in mesothelioma
On the basis of the findings that IGF-I stimulation promotes phosphorylation of both 4E-BP1 and eIF4E, resulting in the inactive and active forms of these proteins, respectively, it was hypothesised that IGF-I stimulation of mesothelioma cells would result in an enhanced eIF4F function and increased translation. To examine the effects of IGF-I stimulation on cap-dependent translation in mesothelioma, synthetic 7mGTP-sepharose beads were used to capture eIF4E and its binding partners, eIF4G and 4E-BP1. Mesothelioma cells were treated with or without IGF-I, and cap-binding assays were performed on cell lysate prepared in the same manner as the assays described in Figure 1. Relatively higher levels of cap-bound eIF4G in cell lysate signify the functional potency of eIF4F, whereas higher levels of cap-captured 4E-BP1 indicate a repression of the eIF4F complex. Consistent with the activation of the cap-mediated translation complex, the quantity of eIF4E-bound eIF4G was increased in most mesothelioma cell lines treated with IGF-I for 20 or 300 min compared with that in untreated cells (Figure 2A, top panel). Furthermore, 4E-BP1 binding decreased substantially (Figure 2A, lowest panel) in most mesothelioma cells treated with IGF-I, indicating an inactivation of 4E-BP1 after phosphorylation. In response to IGF-I stimulation, the ratio of eIF4G to eIF4E was increased 1.5- to 3.5-fold compared with that in untreated cells (Figure 2B). The effect on cap-dependent translation of activating 4E-BP1 by blocking the PI3K/Akt/mTOR-signalling pathway with LY249002 was also examined using the cap-affinity assay. In all mesothelioma cell lines, LY249002 treatment resulted in a decrease in the eIF4G/eIF4E ratio in IGF-I-stimulated cells than in cells treated with only IGF-I (Figure 2B). In some cell lines (H2373, H513, and H2596), the ratio fell to levels at or below that of IGF-I-untreated cells. In addition, the level of eIF4E-bound 4E-BP1 increased in IGF-I stimulated cells treated with LY249002 (Figure 1A), consistent with the suppression of cap-dependent translation.

IGF-I stimulation activates cap-mediated translation in mesothelioma. (A) After overnight serum starvation, cells were treated or not treated with IGF-I (5 nM) for the indicated times (minutes) or treated with IGF-I combined with LY249002 (LY). Samples were subjected to cap-analogue capture using 7-methyl-GTP-sepharose before immunoblot analysis. (B) Depiction of the relative level of eIF4G normalised to eIF4E bound to the cap analogue for each cell line.
Constitutively active 4E-BP1A37/A46 inhibits activation of the eIF4F complex in mesothelioma
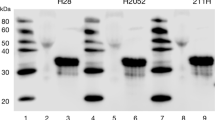
If cap-mediated translation contributes to the malignant phenotype, then repressing its activity should repress the malignant potential of mesothelioma cells. Mesothelioma cells were selected to ectopically express active 4E-BP1 using a phosphorylation-defective 4E-BP1A37/A46. The 4E-BP1A37/A46 construct has substituted alanine for threonine at residues 37 and 46, the first two sites of sequential phosphorylation that render 4E-BP1 incapable of phosphorylation and consequently dominantly active (Gingras et al, 2001). To assess the ability of 4E-BP1A37/A46 to interfere with the assembly of the eIF4F initiation complex, a cap-analogue capture of eIF4E and its binding partners was used, followed by immunoblot analysis. Four mesothelioma cell lines were stably transfected with an empty vector or with the same vector-containing 4E-BP1A37/A46. Expression of exogenously produced 4E-BP1A37/A46 was present in cell lysate for all four cell lines (Figure 3A, top panel). The HA-tagged ectopically expressed 4E-BP1A37/A46 migrates more slowly and is easily monitored (α-HA antibody). For each mesothelioma cell line stably transfected with empty vector, eIF4G avidly bound to eIF4E (Figure 3A, lower panel), which is indicative of a translationally active state. For each cell line, the association of eIF4G to eIF4E was repressed when cells expressed 4E-BP1A37/A46 compared with cells transfected with an empty vector (Figure 3A, lower panel). Formation of the eIF4F initiation complex was inhibited in cells expressing 4E-BP1A37/A46 by 66.7% (H2373) to 51% (H2461) (Figure 3B). Results from the cap-affinity assay show that the quantity of eIF4E bound to 4E-BP1 inversely correlates with the level of eIF4G bound to eIF4E (Figure 3A). The level of the mutually exclusive cap-bound proteins (eIF4G and 4E-BP1) indicates that ectopically expressed 4E-BP1A37/A46 strongly diminishes the activation of the cap-dependent complex.

Assembly of cap-dependent initiation complex in mesothelioma is impaired by the production of constitutively active 4E-BP1 (4E-BP1A37/A46). (A) An assessment of eIF4F integrity utilising a cap-affinity assay of four mesothelioma cell lines ectopically expressing the 4E-BP1A37/A46 protein or control. Top, steady state levels of exogenous HA-tagged 4E-BP1A37/A46 and endogenous 4E-BP1 in cells. Bottom, eIF4E and binding partners eIF4G and 4E-BP1A37/A46 eluted from 7-methyl-GTP-sepharose resin. (B) The average and s.d. for the 4 MM cell lines, relative level of eIF4G normalised to eIF4E bound to the cap analogue comparing cells expressing and not expressing 4E-BP1A37/A46.
Constitutively active 4E-BP1A37/A46 inhibits IGF-I-mediated effects on mesothelioma
The previous results demonstrate that IGF-I stimulation of mesothelioma cells directly promotes activation of the eIF4F complex. The next step was to evaluate the link between cap-mediated translation and mesothelioma cell cloning efficiency, proliferation, motility, and tumourigenicity. Each of the four mesothelioma cell lines was transfected with either the dominant active 4E-BP1A37/A46 gene or the empty vector. Colonies were counted and normalised to that of the empty vector. In all four mesothelioma cell lines, ectopically expressed 4E-BP1A37/A46 exhibited a dramatic impact on cloning efficiency (Figure 4).

Constitutively active 4E-BP1 hinders mesothelioma colony formation. Mesothelioma cell lines were transfected with empty vector (open columns) or HA-4E-BP1A37/A46 (filled columns). G418-resistant clones were counted for each transfection and normalised to the value for empty vector transfection for each cell line.
To assess the effects on motility and proliferation when cap-mediated translation is repressed, mesothelioma cell lines were stably transfected with empty vector or with the 4E-BP1A37/A46 dominant active gene. Transfected cells and control parental cells were grown in SFM in the presence or absence of IGF-I (Figure 5). In both the parental and empty vector-containing cell lines, IGF-I exposure led to increased proliferation compared with that in untreated cells (Figure 5). 4E-BP1A37/A46 expression resulted in diminished proliferation. In a similar manner, 4E-BP1A37/A46 expression decreased mesothelioma cell motility after IGF-I stimulation (Figure 6), although the results did not reach statistical significance. Parental and empty vector-containing H2461 cells showed a 4- and 2.6-fold enhancement of cell migration, respectively, when exposed to IGF-I than did untreated cells. In contrast, the migration of H2461 cells expressing 4E-BP1A37/A46 did not increase in response to IGF-I stimulation. These results demonstrate that the proliferative and migratory responses of mesothelioma cells after IGF-I treatment are blunted by repressing cap-mediated translation.

Abrogation of IGF-I-stimulated cell proliferation by a constitutively active 4E-BP1. H2373 and H2461 cell lines expressing dominant active 4E-BP1A37/A46 in the presence or absence of IGF-I (5 nM), and 24 h later, cell growth was measured. Columns, the mean of three independent determinations of cell number, bars, s.d.

Suppression of IGF-I activated cell motility by disruption of cap-mediated translation. H2461 cells that do and do not produce dominant active 4E-BP1A37/A46 in the presence (filled columns) or absence (open columns) of IGF-I (5 nM). After 24 h incubation, cell migration was assayed. Columns, the mean of three independent determinations of migrating cells, bars, s.d.
Constitutively active 4E-BP1A37/A46 abrogates xenograft tumour growth
The IGF-I stimulation of cap-mediated translation in mesothelioma is ultimately only important if it contributes to the tumourigenic potential of mesothelioma cells. To demonstrate the relationship between cap-mediated translation and in vivo tumour formation, H2596 or H2373 cells ectopically expressing 4E-BP1A37/A46 or empty vector were injected into nude mice. Animals were killed, tumours were excised, and weighed on days 14 and 27 after implantation of H2596 and H2373 cells, respectively. Expression of 4E-BP1A37/A46 caused a marked suppression of tumour growth in these mesothelioma xenografts (Figure 7B). H2373 cells expressing 4E-BP1A37/A46 produced tumours that were one-third the size of parental controls. H2596 cells expressing 4E-BP1A37/A46 did not produce tumours in any mice. Lysate prepared from H2373 cells expressing empty vector or 4E-BP1A37/A46 was subjected to immunoblot analysis both before and after implantation into mice (Figure 7A). The level of expression of the 4E-BP1A37/A46 protein diminished during xenograft tumour growth to a nearly undetectable level in H2373 xenograft tumours, suggesting that tumour growth occurred after the selection of cells expressing minimal levels of 4E-BP1A37/A46. These results show that suppression of aberrant cap-dependent translation in mesothelioma cells abrogates tumour growth.

Xenograft tumour growth is abrogated by constitutively active 4E-BP1. (A) tumour size for 10 animals (mean±s.e.) from cell lines H2373 and H2596 expressing (filled columns) or not expressing (open columns) 4E-BP1A37/A46. Columns, the mean of five xenograft measurements of each cell line, bars, s.e. (B) immunoblot analysis of lysates from mesothelioma cells producing or not producing 4E-BP1A37/A46 both before (in vitro) and after implantation (xenograft) into the mice. The level of ectopic HA-tagged 4E-BP1 is shown.
Discussion
Activated cap-mediated protein translation is found in many cancers (Mamane et al, 2004). This activation is associated with the preferential translation of a limited number of transcripts responsible for cell proliferation and survival (Graff and Zimmer, 2003; Rajasekhar et al, 2003). Enhanced cap-mediated translation in cancer often occurs after either an overexpression of eIF4E, phosphorylation of eIF4E, or an inactivation of the 4E-BP1 repressor protein. In mesothelioma, the primary mechanism for elevated levels of cap-mediated translation seems to be a low-level expression of the 4E-BP1 repressor protein in conjunction with an activation of eIF4E (Patel et al, 2007). This is in contrast to previous observations in cancers, such as non-small-cell lung cancer, in which both elevated levels of eIF4E and inactivation of the 4E-BP1 repressor protein are the primary mechanisms for activating the eIF4F complex (Jacobson et al, 2006). This study shows that IGF-I stimulation of mesothelioma cells results in a similar hyperphosphorylation and inactivation of 4E-BP1 (Figure 1).
It is well known that IGF-I stimulation of eukaryotic cells can lead to increased protein translation and contribute to the malignant potential in a variety of cell types (Miller and Yee, 2005). In mesothelioma, IGF-I stimulation leads to the downstream activation of both PI3K/Akt and MAPK pathways (Hoang et al, 2004b). The observation that IGF-I stimulation is associated with increased phosphorylation of 4E-BP1, allowing for an increase in the binding of the competing eIF4G protein, thus favouring protein translation, is not unexpected and is consistent with IGF-I-driven malignant growth in mesothelioma. In support of the function of the IGF-I axis in mesothelioma development, previous data demonstrate that malignant transformation of mesothelial cells in animal models requires an intact IGF1R molecule (Pass et al, 1996). An intact and active IGF-I axis is necessary for both the development and propagation of mesothelioma cells.
Activation of IGF1R is associated with an increase in both mesothelioma cell proliferation and motility (Hoang et al, 2004b). In this study, when cap-mediated protein translation was suppressed using the dominantly active mutant protein, 4E-BP1A37/A46, mesothelioma cells showed diminished proliferation and motility even under conditions of IGF-I stimulation. IGF-I stimulation of mesothelioma cells also led directly to the activation of the cap-mediated translation complex. The importance of activation or repression of cap-mediated translation in mesothelioma is strongly emphasised by the observation that an enforced repression of translation in mesothelioma cells resulted in the near total loss of tumourigenic potential.
Our finding that cap-mediated protein translation is associated with IGF-I stimulation suggests that IGF1R inhibition is a very attractive therapeutic target in mesothelioma. IGF1R inhibitors are currently in clinical testing and seem to have potent in vitro activity against mesothelioma cells (Whitson et al, 2006). There are data that these agents are synergistic with cytotoxic chemotherapy, an observation that fits well with the pro-apoptotic effects known to accompany a repression of cap-mediated translation. We have previously shown that an enforced repression of cap-mediated activity can render cancer cells more susceptible to chemotherapeutic-induced apoptosis in non-small-cell lung cancer cells (Jacobson et al, 2006). Suppressing IGF1R activation should have the same effect of reducing cap-mediated translation and enhancing chemosensitivity. In addition, the IGF1R-mediated activation of PI3K leads to an acquired resistance to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, in A431 lung cancer cells (Guix et al, 2008). The inhibition of both EGFR and IGF1R was required to reverse acquired in vitro drug resistance. It is tempting to speculate that both oncogenic pathways converge upon the initiation of translation to mediate their oncogenic effects. If so, the initiation of cap-dependent translation would be an attractive target for new therapies, as it would potentially block the downstream signal of two major oncogenic pathways.
Novel agents are being developed for targeting eIF4E or for disabling the eIF4F molecular complex as a potential cancer therapy (Graff et al, 2007, 2008). On the basis of these data demonstrating the important function that IGF-I stimulation and cap-mediated protein translation have in the malignant potential of mesothelioma cells, these therapies could prove to be promising treatments for mesothelioma. In the light of the findings that the effects of IGF1R activation in mesothelioma depend on the activation of the eIF4F complex, it will be interesting to combine IGF1R inhibition with agents designed to target cap-mediated translation.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Britton M (2002) The epidemiology of mesothelioma. Semin Oncol 29: 18–25
De Benedetti A, Graff JR (2004) eIF-4E expression and its role in malignancies and metastases. Oncogene 23: 3189–3199
Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N (2001) Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15: 2852–2864
Graff JR, Konicek BW, Carter JH, Marcusson EG (2008) Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res 68: 631–634
Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, Cox K, McNulty AM, Parsons SH, Wang T, Sams L, Geeganage S, Douglass LE, Neubauer BL, Dean NM, Blanchard K, Shou J, Stancato LF, Carter JH, Marcusson EG (2007) Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest 117: 2638–2648
Graff JR, Zimmer SG (2003) Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis 20: 265–273
Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA (2008) Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 118: 2609–2619
Haddad T, Yee D (2006) Targeting the insulin-like growth factor axis as a cancer therapy. Future Oncol 2: 101–110
Hoang CD, D’Cunha J, Kratzke MG, Casmey CE, Frizelle SP, Maddaus MA, Kratzke RA (2004a) Gene expression profiling identifies matriptase overexpression in malignant mesothelioma. Chest 125: 1843–1852
Hoang CD, Zhang X, Scott PD, Guillaume TJ, Maddaus MA, Yee D, Kratzke RA (2004b) Selective activation of insulin receptor substrate-1 and -2 in pleural mesothelioma cells: association with distinct malignant phenotypes. Cancer Res 64: 7479–7485
Jacobson BA, Alter MD, Kratzke MG, Frizelle SP, Zhang Y, Peterson MS, Avdulov S, Mohorn RP, Whitson BA, Bitterman PB, Polunovsky VA, Kratzke RA (2006) Repression of cap-dependent translation attenuates the transformed phenotype in non-small cell lung cancer both in vitro and in vivo. Cancer Res 66: 4256–4262
Jerome L, Shiry L, Leyland-Jones B (2003) Deregulation of the IGF axis in cancer: epidemiological evidence and potential therapeutic interventions. Endocr Relat Cancer 10: 561–578
Kratzke RA, Otterson GA, Lincoln CE, Ewing S, Oie H, Geradts J, Kaye FJ (1995) Immunohistochemical analysis of the p16INK4 cyclin-dependent kinase inhibitor in malignant mesothelioma. J Natl Cancer Inst 87: 1870–1875
Lee AY, Raz DJ, He B, Jablons DM (2007) Update on the molecular biology of malignant mesothelioma. Cancer 109: 1454–1461
Lee TC, Zhang Y, Aston C, Hintz R, Jagirdar J, Perle MA, Burt M, Rom WN (1993) Normal human mesothelial cells and mesothelioma cell lines express insulin-like growth factor I and associated molecules. Cancer Res 53: 2858–2864
Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N (2004) eIF4E--from translation to transformation. Oncogene 23: 3172–3179
Miller BS, Yee D (2005) Type I insulin-like growth factor receptor as a therapeutic target in cancer. Cancer Res 65: 10123–10127
Pass HI, Mew DJ, Carbone M, Matthews WA, Donington JS, Baserga R, Walker CL, Resnicoff M, Steinberg SM (1996) Inhibition of hamster mesothelioma tumorigenesis by an antisense expression plasmid to the insulin-like growth factor-1 receptor. Cancer Res 56: 4044–4048
Patel MR, Jacobson BA, De A, Frizelle SP, Janne P, Thumma SC, Whitson BA, Farassati F, Kratzke RA (2007) Ras pathway activation in malignant mesothelioma. J Thorac Oncol 2: 789–795
Polunovsky VA, Bitterman PB (2006) The cap-dependent translation apparatus integrates and amplifies cancer pathways. RNA Biol 3: 10–17
Prevot D, Darlix JL, Ohlmann T (2003) Conducting the initiation of protein synthesis: the role of eIF4G. Biol Cell 95: 141–156
Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC (2003) Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 12: 889–901
Sachdev D, Yee D (2006) Inhibitors of insulin-like growth factor signaling: a therapeutic approach for breast cancer. J Mammary Gland Biol Neoplasia 11: 27–39
Thumma SC, Kratzke RA (2007) Translational control: a target for cancer therapy. Cancer Lett 258: 1–8
Whitson BA, Jacobson BA, Frizelle S, Patel MR, Yee D, Maddaus MA, Kratzke RA (2006) Effects of insulin-like growth factor-1 receptor inhibition in mesothelioma. Thoracic Surgery Directors Association Resident Research Award. Ann Thorac Surg 82: 996–1001; discussion 1001–2
Whitson BA, Kratzke RA (2006) Molecular pathways in malignant pleural mesothelioma. Cancer Lett 239: 183–189
Workman P, Twentyman P, Balkwill F, Balmain A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, Stables J, Stephens T, Wallace J (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the welfare of animals in experimental neoplasia (second edition). Br J Cancer 77: 1–10
Acknowledgements
We thank Douglas Yee and Deepali Sachdev for their helpful advice and comments. We thank Michael Franklin for editorial assistance with this article. This work is supported in part by a grant from the Mesothelioma Applied Research Foundation (RAK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Jacobson, B., De, A., Kratzke, M. et al. Activated 4E-BP1 represses tumourigenesis and IGF-I-mediated activation of the eIF4F complex in mesothelioma. Br J Cancer 101, 424–431 (2009). https://doi.org/10.1038/sj.bjc.6605184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605184
Keywords
This article is cited by
-
4Ei-10 interdiction of oncogenic cap-mediated translation as therapy for non-small cell lung cancer
Investigational New Drugs (2021)
-
Repression of oncogenic cap-mediated translation by 4Ei-10 diminishes proliferation, enhances chemosensitivity and alters expression of malignancy-related proteins in mesothelioma
Cancer Chemotherapy and Pharmacology (2020)
-
Inhibition of oncogenic cap-dependent translation by 4EGI-1 reduces growth, enhances chemosensitivity and alters genome-wide translation in non-small cell lung cancer
Cancer Gene Therapy (2019)
-
4EGI-1 represses cap-dependent translation and regulates genome-wide translation in malignant pleural mesothelioma
Investigational New Drugs (2018)
-
Multipoint targeting of the PI3K/mTOR pathway in mesothelioma
British Journal of Cancer (2014)
