
Abstract
Vascular endothelial growth factor-A is widely regarded as the principal stimulator of angiogenesis required for tumour growth. VEGF is generated as multiple isoforms of two families, the pro-angiogenic family generated by proximal splice site selection in the terminal exon, termed VEGFxxx, and the anti-angiogenic family formed by distal splice site selection in the terminal exon, termed VEGFxxxb, where xxx is the amino acid number. The most studied isoforms, VEGF165 and VEGF165b have been shown to be present in tumour and normal tissues respectively. VEGF165b has been shown to inhibit VEGF- and hypoxia-induced angiogenesis, and VEGF-induced cell migration and proliferation in vitro. Here we show that overexpression of VEGF165b by tumour cells inhibits the growth of prostate carcinoma, Ewing's sarcoma and renal cell carcinoma in xenografted mouse tumour models. Moreover, VEGF165b overexpression inhibited tumour cell-mediated migration and proliferation of endothelial cells. These data show that overexpression of VEGF165b can inhibit growth of multiple tumour types in vivo indicating that VEGF165b has potential as an anti-angiogenic, anti-tumour strategy in a number of different tumour types, either by control of VEGF165b expression by regulation of splicing, overexpression of VEGF165b, or therapeutic delivery of VEGF165b to tumours.
Similar content being viewed by others


Main
Vascular endothelial growth factor (VEGF-A) is the principal angiogenic promoter in most, if not all, cancers acting primarily on endothelial cells through its cognate receptors VEGF-R1 and VEGF-R2. VEGF is upregulated by hypoxia (Shweiki et al, 1992), and by overexpression of oncogenes such as mutant ras (Rak et al, 1995) and c-myc (Mezquita et al, 2005) in tumours, and stimulates the migration of endothelial cells, sprouting of blood vessels, and generation of new vessels from existing vasculature in tumours (reviewed in Ferrara, 2004). The resulting sustained blood flow, oxygen supply and waste removal enables more rapid growth of the tumour. Anti-VEGF therapy is now an additional therapeutic strategy to surgery, chemotherapy and radiotherapy, and recent trials of antibodies to VEGF as adjuvant therapy have shown significant clinical benefit in colorectal cancer (Hurwitz et al, 2004), renal carcinoma, non small cell lung, ovarian and other cancers.
Vascular endothelial growth factor is generated as multiple isoforms by alternative splicing of mRNA from 8 exons (Houck et al, 1991). Conventional VEGF isoforms are pro-angiogenic, pro-permeability vasodilators. These isoforms contain exons 1–5 and 8a, with a variable contribution from exons 6a, 6b, 7a, and 7b, resulting in a family of peptides identified numerically by their amino-acid content, each with different heparin-binding properties (Houck et al, 1991). These are generically identified as VEGFxxx, where xxx refers to the number of amino acids. In 2002 (Bates et al, 2002) and 2004 (Woolard et al, 2004) we identified a sister family of isoforms of identical lengths and exon structure apart from the C-terminal exon in which distal splicing results in an alternate open-reading frame of six amino acids (exon 8b rather than 8a, SLTRKD vs CDKPRR) – generically referred to as VEGFxxxb (e.g. VEGF165b, VEGF121b (Perrin et al, 2005), VEGF189b (Miller-Kasprzak and Jagodzinski, 2008)). These isoforms are antiangiogenic and downregulated in renal tumours and metastatic melanoma (Bates et al, 2002; Pritchard-Jones et al, 2007). This antiangiogenic activity, generated by receptor binding but only weak receptor activation (Cebe Suarez et al, 2006) and inhibition of downstream VEGF-R2 signalling, has led to the hypothesis that VEGF165b, or manipulation of C-terminal VEGF splicing to enhance more distal splicing, may be useful therapeutic tools in cancer.
We have previously shown that VEGF165b is present in a range of normal tissues (Bates et al, 2002; Woolard et al, 2004) and there is a downregulation of the antiangiogenic VEGF165b protein in malignant prostate cancer (Woolard et al, 2004) and metastatic melanoma (Pritchard-Jones et al, 2007) and mRNA in renal carcinoma (Bates et al, 2002). We also showed that VEGF165 expressing tumours grew faster than VEGF165b expressing tumours, suggesting that VEGF165b was not angiogenic. Moreover, we showed that tumours in which VEGF165b expressing cells were mixed with VEGF165 expressing cells grew more slowly than cells only expressing VEGF165 (but more quickly than cells expressing VEGF165b alone). This latter finding rules out a possibility that VEGF165b could be angiogenic in the presence of VEGF165. However, neither that, nor any other study, has shown whether VEGF165b can inhibit tumour growth, whether it can inhibit VEGF165-mediated tumour growth, or whether it can do so by inhibiting angiogenesis.
In this study, we have examined the effect of VEGF165b overexpression on tumour growth, and VEGF165-mediated angiogenesis of prostate and renal cell carcinoma and on growth of Ewing's sarcoma cell and metastatic melanoma in xenografted mouse models.
Materials and methods
Human tissue and RT–PCR of human TURP chips
Frozen prostate chips were obtained from patients undergoing transurethral resection of the prostate (TURP) for lower urinary tract symptoms with benign prostatic hyperplasia and advanced prostate cancer (stage T3 Nx M0-1; UICC2002). The use of tissue was approved by the NBHST Ethical Committee.
Fifty to 100 mg of 26 TURP tissue (nine malignant, 17 benign prostatic hypertrophy) was homogenised in Trizol reagent (Life Technologies Inc., Rockville, MD, USA) and mRNA was extracted by using the method of (Chomczynski and Sacchi, 1987). Eight microliters of RNA were treated with RNase free DNase (Promega, Madison, WI, USA) according to the manufacturer's guidelines to prevent genomic DNA contamination and mRNA was reverse transcribed using Moloney murine leukaemia virus reverse transcriptase and poly-d(T). cDNA was then amplified using intron spanning primers that detect VEGF165b only, even in the presence of 1000 fold greater concentration of VEGF165 mRNA (Bates et al, 2002). The cDNA was amplified using 1 μ M intron-spanning primers designed to detect VEGFxxxb (Bates et al, 2002) (Exon 4, 5′-GAGATGAGCTTCCTACAGCAC-3′ and (Exon8b/7, 5′-TTAAGCTTTCAGTCTTTCCTGGTGAGAGATCTGCA-3′) or VEGFxxx with exon 8a (5′-CACCGCCTCGGCTTGTCACAT-3′), together with 1.2 mM MgCl2, 200 μ M dNTPs, and 1 unit of Taq DNA polymerase (Abgene), cycled 35 times, at 94°C for 30 s, 63°C for 30 s and 72°C for 60 s. β2-microglobulin was used as control amplification (β2 forward primer 5′-GCATCATGGAGGTTTGAAGATG-3′, β2 reverse 5′-TAAGTTGCCAGCCCTCCTAGAG-3′) at 55°C annealing temperature leading to a 220 bp product. Full-length VEGF165b or VEGF165 in pcDNA3 vector were used as positive and negative controls. PCR products were run on 3% agarose gels. PCR bands were excised and extracted using Qiaex (Qiagen, Crawley, UK), cloned using a TOPO TA Cloning® Kit (Invitrogen, Paisley, UK) and confirmed by sequencing.
Establishment of overexpressing tumour cells
All media and supplements were from Gibco/Invitrogen if not otherwise stated. PC3 cells were kindly donated by Professor J Masters from UCH, University of London. Ewing's sarcoma cell line, TC71, renal cell carcinoma cell line, CAKI, prostate cell line, PC3, and melanoma cell line, Mel57, were grown in Eagle's modified essential medium supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids, NEAA, 2% minimum essential medium vitamin solution, minimum essential media (Sigma Aldrich, Dorset, UK) supplemented with 10% FCS, 1% L-glutamine, 1% penicillin/streptomycin, 1% NEAA or McCoy's 5A medium supplemented with 10% FCS and Ham F-12 supplemented with 10% FCS, 1% penicillin/streptomycin, 1.5 g l−1 sodium bicarbonate (Sigma Aldrich), respectively.
For transfection, 90% confluent cells in 6-well plates were transfected with 0.5 μg of plasmid DNA (pcDNA empty vector, pcDNA-VEGF165, pcDNA-VEGF165b or equal amounts of VEGF165 and VEGF165b plasmids) using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to manufacturers protocols and Optimem media. Cells were selected with 375 μg ml−1 of geneticin and maintained at 125 μg ml−1. Transfected cells were analysed by an in-house developed VEGF165b-specific enzyme-linked immunosorbent assay (ELISA) (Perrin et al, 2005) or a commercial panVEGF ELISA (Duoset, R&D Systems), that detects all isoforms of VEGF-A, to validate expression levels in selected cells. ELISA was performed either according to manufacturers instructions or as previously described (Woolard et al, 2004). Conditioned media from 200 000 cells was collected and was analysed by ELISA after 48 h incubation. Mel57 cells were transfected with plasmid pIRESneo containing the VEGF165 or VEGF165b cDNA essentially as described before (Kusters et al, 2007).
Migration of cells with conditioned media
Cultured supernatants from TC71, empty vector transfected TC71 or TC71/VEGF165b cells were collected. Transwells (Costar, Cambridge, MA, USA) were pretreated with serum-free medium at 37°C for 1 h before seeding with human dermal microvascular endothelial cells, (HMVECs)at 1 × 105 per well in 100 μl endothelial basal media (Cambrex, Baltimore, MD, USA) with 0.1% fetal bovine serum. These cells are commercially available cells generated from human foreskin tissue. The transwells were then inserted into 24-well plates containing 600 μl of conditioned medium and incubated at 37°C for 6 h to allow HMVEC cells to migrate. Cells on the upper side of the filter were removed with cotton swabs. Migrated cells on the lower side of the filter were fixed and stained with haematoxylin and eosin. The number of migrated cells was counted under a binocular microscope.
[3H]-thymidine incorporation of cells with conditioned media
Human dermal microvascular endothelial cells (3 × 103) were seeded into 96-well cell culture plates and allowed to adhere for 5 h before the addition of conditioned medium from TC71 cells, empty vector transfected TC71 cells or TC71/VEGF165b cells. Triplicate wells were used for each group. The cultures were labelled with 0.2 μCi per well of [3H] thymidine (ICN Biomedicals Inc., Radiochemicals Division, Irvine, CA, USA) during the last 24 h of a 48 h incubation. At the end of incubation, the cells were washed two times with HBSS, and 0.1 ml of 0.1 N KOH was added to lyse the adherent cells. The radioactive incorporation was determined using a plate harvester (Brandal Biomedical Research and Development Lab Inc., Gaithersburg, MD, USA) and Beta Plate Counter (Wallac 1450 Micro beta Counter, Perkin Elmer Life Sciences, Turku, Finland).
In vitro growth analysis of cells
For direct counting and proliferation of transfected CAKI cells, 30 000 cells per 24 well were seeded out in triplicates. Cells were maintained in media supplemented with 0.01% or 10% FCS and were counted after 24 or 48 h. The doubling time for each cell population was calculated using Prism software. The metabolic rate was analysed by seeding 5000 cells per 96 well in media supplemented with 0.01% FCS. After 24 or 48 h 50 μl of metabolic reagent was added (CellTiter 96® Aqueous One solution, Promega, Madison, WI, USA). Cells were incubated for 4 h at 37°C and analysed at 490 nm.
Animal housing and xenograft model
One million Ewing sarcoma or Mel57, 8 million CAKI, 3 million PC3 overexpressing cells were injected subcutaneously into the back of unanaesthetised nude mice in 100 μl sterile PBS. Xenotransplanted tumours were measured by calliper and tumour volume was calculated according to (length × width × (length+width)/2). Mice were culled by cervical dislocation when tumours reached 16 mm in any direction and organs and tumours were removed and snap-frozen or fixed in 4% PFA followed by paraffin embedding and 5 μm sections were generated and stained with CD31/PECAM. Slides were boiled for 10 min in 0.01 M sodium citrate boiling, followed by blocking in 5% goat serum for 1 h, overnight incubation with 2.5 μg ml−1 anti-mouse PECAM antibody (Pierce Endogen) and for 1 h in 5 μg ml−1 ALEXA Fluor 488 goat anti hamster IgG (Invitrogen, Molecular Probes). Vessels were counted in six different fields at × 20 magnification and verified at × 40 magnification. Nude mice were kept under appropriate specific pathogen-free housing facilities according to government guidelines and procedures were carried out according to national guidelines and regulations.
Statistical analysis
Statistical analysis was performed using Prism. All data are given as mean±s.e.m. if not otherwise stated. Number of benign TUR and malignant chips expressing VEGF165b was compared using Fisher's exact test. For tumour growth one-way analysis of variance (ANOVA) followed by Newman–Keuls' post hoc test (prostate) or two-way ANOVA followed by Bonferroni post hoc test (renal carcinoma) were used as data allowed. Weight, blood score, proliferation, cell doubling and metabolic rate were analysed by one-way ANOVA followed by Newman–Keuls' multiple comparison post hoc test. P<0.05 was considered significant.
Results
VEGF165b is downregulated in malignant prostate tumours compared to benign prostate tissue and reduced tumour growth in xenograft model
RNA was extracted from human prostate chips from patients undergoing TURP and expression of VEGF165 and VEGF165b mRNA was analysed by isoform specific PCR. Expression of VEGF165b was found in 10 out of 11 benign samples and VEGF165 was also present in 10 out of 11 (see Figure 1A). In malignant prostate on the other hand, VEGF165b was only found in four out of nine, whereas the VEGF165 expression was found in eight out of nine (see Figure 1B).

The antiangiogenic VEGF165b is downregulated in human malignant prostate tumour and reduces prostate tumour growth. Anonymous examples of VEGFxxx and VEGFxxxb mRNA expression in TURP chips from benign prostate hypertrophy (A) and malignant prostate cancer (B). (A) RT–PCR of mRNA extracted from benign prostate chips using primers to detect VEGFxxxb and VEGFxxx. Ten out of 11 benign samples showed expression of VEGFxxxb. All but one sample also showed VEGF165 expression. (B) RT–PCR of RNA extracted from malignant prostate chips. Only four of the nine malignant samples showed expression of VEGF165b and eight out of nine malignant samples showed VEGF165 expression. β-microglobulin expression was seen in all the malignant tissues (data not shown). (C–F) Images of tumour-bearing mice. Tumours from PC3 cells overexpressing empty pcDNA3 vector (C), VEGF165 (D), VEGF165b (E) and cotransfection with VEGF165 and VEGF165b (F). Scale bar=10 mm. (G) VEGF165b reduced prostate tumour growth in a xenograft mouse model. Three million PC3 cells were injected and VEGF165b overexpression reduced control and VEGF165-mediated tumour growth at day 18 (P<0.05 Kruskal–Wallis, *control vs VEGF165b P<0.05, +VEGF165 vs VEGF165b P<0.05).
Human prostate cancer cells, PC3 cells, were transfected with control, VEGF165b, VEGF165 expression plasmids, or a combination of the two and injected into nude mice. The tumour volume was monitored over time and at day 18 the tumour volumes were significantly different (P<0.05). Figure 1C–F illustrates representative images of each group. The pro-angiogenic VEGF165 transfected cells produced rapidly growing tumours although not statistically different from control tumours (P>0.5). Conversely the VEGF165b transfectants resulted in smaller tumours (P<0.05 at 22 days compared with pcDNA3 controls), and those expressing both isoforms were significantly smaller than those expressing VEGF165 only (P<0.05 at 18 days).
VEGF165b overexpression reduces renal cell carcinoma growth
Renal carcinoma (CAKI) cells, transfected with the different VEGF isoforms were analysed for VEGF expression. VEGF165b was undetectable in empty vector and VEGF165-transfected cells and VEGF165b expression was readily detectable in the VEGF165b- or cotransfected cells (Table 1). The endogenous levels of VEGF165 were high in CAKI cells and transfection of VEGF165 increased the total levels (Table 1).
Eight million transfected CAKI cells were injected into nude mice and monitored over time. Injection of control transfected cells resulted in solid, bloody tumours after 29 days (see Figure 2A), and VEGF165-expressing tumours resulted in larger, bloody tumours after 29 days (see Figure 2B), VEGF165b-expressing tumours, while they did grow (see Figure 2C) were smaller, and less bloody. Cells expressing both isoforms also resulted in small, relatively blood-free tumours that were not different from the VEGF165b-expressing tumours (see Figure 2D). Figure 2E shows that VEGF165b-expressing tumours grew significantly slower than VEGF165-expressing tumours (overall P<0.001, VEGF165 vs VEGF165b P<0.05 at day 27, VEGF165 vs both P<0.05 day 24 and P<0.001 day 27). No significant difference was found between tumours formed from cells expressing VEGF165 and control or between those expressing VEGF165b and both VEGF isoforms.

VEGF165b inhibits tumour growth in renal cell carcinomas. Renal cell carcinoma (8 × 106 CAKI) cells, transfected with pcDNA3 vectors expressing empty vector (A) VEGF165 (B), VEGF165b (C) or cotransfected with VEGF165 and VEGF165b (D) were injected into the back of nude mice, n=6 mice per group, photographs taken at 29 days. Tumour border outlined by dotted line. Inset shows excised tumours. (E) Tumour growth curves. (F) Tumour weight at day of culling. (G) Macroscopic estimation of blood content at day of culling. (*P<0.05 VEGF165b vs VEGF165, ΔP<0.05 ΔΔΔP<0.001 VEGF165+165b vs VEGF165, ##P<0.01 VEGF165 vs pcDNA).
Upon excision of the tumours, they were weighed and scored blindly for blood content. VEGF165-expressing tumours were significantly larger (see Figure 2F) than control and VEGF165b-expressing tumours, which also resulted in a significant reduction of tumour volume (P<0.05 VEGF165 vs both isoforms or VEGF165b). There was a non-significant reduction in tumour volume for VEGF165b-expressing tumours compared with control transfected. Macroscopically the tumours differed with more blood in VEGF165-expressing tumours compared to any other of the groups (P<0.01, VEGF165 vs either of the other groups, Figure 2G). Similar data was observed with a smaller initial tumour cell injection (2 × 106 cells, data not shown).
The growth rates of the transfected CAKI cells were analysed. There was no difference in the proliferation as measured by direct counting of cells (see Figure 3A and B) or analysis of metabolic rate (see Figure 3C). Both experiments were performed in the presence of serum with the same results (data not shown).

Overexpression of VEGF165b does not affect proliferation when overexpressed in renal cell carcinoma cells in vitro. (A, B) Transfected renal cell carcinoma CAKI cell growth was analysed by direct counting of cells after 24 and 48 h in low serum (0.01% FCS). The doubling time was calculated for each cell population. No significant differences were observed in either instance (P>0.05 n=3, one-way ANOVA). (C) Cell viability and metabolic rate was analysed at 48 h in low serum with transfected CAKI cells. No significant difference was observed (P>0.05 n=4, one-way ANOVA).
These results indicate that VEGF165b overexpression reduced tumour growth in renal cell carcinoma cells grown in mice even in the presence of VEGF165. The tumour inhibition by VEGF165b appears not to be through reduction in tumour cell proliferation.
VEGF165b overexpression reduced Ewing's sarcoma growth and tumour-conditioned media reduced endothelial cell proliferation and migration
Injection of TC71 Ewing's sarcoma cells overexpressing VEGF165b subcutaneously into the back of nude mice resulted in tumours that grew significantly slower than control cells over a time period of 29 days (see Figure 4A). To determine whether VEGF165b secreted from the tumour cells was active on the cells that go on to form blood vessels, conditioned media from the Ewing's sarcoma cells, TC71 cells, overexpressing VEGF165b was used to study migration of human microvascular vein endothelial cells, HMVEC. Fetal calf serum resulted in migration of HMVEC as expected (see Figure 4B), and conditioned media from control-transfected tumour cells induced a similar level of migration of HMVEC (see Figure 4C). VEGF165b overexpressing tumour cells resulted in less migration compared to tumour-conditioned media or control-stimulated cells (see Figure 4D).

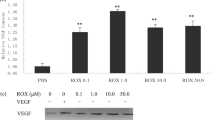
VEGF165b transfection reduces migration, proliferation and tumour growth in vivo of Ewing's sarcoma tumours. (A) VEGF165b overexpression in Ewing's sarcoma cells resulted in significantly smaller tumours 30 days after implantation of 1 × 106 cells, P<0.05 after 7 days, one way ANOVA. (B) Human microvascular endothelial cells, HMVECs (stained with haematoxylin), migrated towards 10% serum and to conditioned media from Ewing's sarcoma cells (C). In contrast VEGF165b overexpression by these cells reduced migration compared to conditioned media and 10% serum (D). (E) When HMVECs were incubated in conditioned media from tumour cells VEGF165 (100 ng ml−1) could still stimulate increased proliferation. Conditioned media from cells overexpressing VEGF165b inhibited this increase.
To determine whether VEGF165b could inhibit growth of endothelial cells, DNA synthesis in HMVEC was measured. VEGF165 increased [3H]-incorporation into HMVECs when added to control tumour cell-conditioned medium. VEGF165b-conditioned media did not increase DNA synthesis of HMVEC cells and reduced VEGF165-mediated DNA synthesis (see Figure 4E). However, VEGF165b did not reduce proliferation below the level of control-conditioned media, indicating that the inhibition of proliferation was specific for VEGF165-mediated proliferation.
Rate of tumour growth depends upon the VEGF isoform expression
The results above suggest that switching expression of VEGF from VEGF165 to VEGF165b might result in reduced tumour growth rates. To determine whether this was the case a VEGF-deficient melanoma cell line (Mel57), which normally grows slowly by co-option of existing vasculature (Westphal et al, 2000) was transfected with VEGF165 or VEGF165b, and implanted subcutaneously. Figure 5 shows that whereas VEGF165-expressing tumours rapidly grew, VEGF165b expressing-tumours were very slow growing, matching that of the previously published co-option dependent parental cell line, indicating that switching expression from VEGF165 to VEGF165b by altering splicing may be a useful therapeutic strategy.

Switching expression from VEGF165 to VEGF165b inhibits tumour growth. Mel57 melanoma cells, which express very low levels of VEGF in vivo were transfected with VEGF165 or VEGF165b and 1 × 106 cells injected subcutaneously into nude mice. Whereas the VEGF165 transfected cells grew rapidly, VEGF165b transfected cells grew no more quickly than previous studies have shown for this VEGF-deficient cell type.
VEGF165b inhibits VEGF165-mediated tumour vessel in-growth
To examine the mechanism for the reduction of tumour growth in vivo and the proposed anti-angiogenic effect of VEGF165b, tumour vessels were visualised by PECAM staining of excised tumour sections. In the CAKI tumours there was a significant increase in microvascular density from 7.03±0.86 per high power field in control (Figure 6A) to 8.37±1.06 in VEGF165-expressing tumours (Figure 6B). In contrast, there was a significant reduction in MVD to 2.17±0.65/hpf in VEGF165b-expressing tumours (P<0.01 compared with control Figure 6C), indicating that VEGF165b inhibited vessel growth in CAKI tumours. This was also significantly lower than VEGF165 (P<0.001). Furthermore, tumours in which VEGF165b and VEGF165 were coexpressed also had significantly reduced MVD (2.98±0.56, Figure 6D, P<0.001 compared with VEGF165), indicating that VEGF165b inhibited VEGF165-mediated blood vessel growth (Figure 6E). Similar results were seen in PC3 cells (Control 8.13±0.40, VEGF165 7.17±1.66, VEGF165b 0.78±0.78, VEGF165+VEGF165b 0.64±0.64. see Figure 6F–J).

VEGF165b reduces vessel density in tumours. Tumour sections were stained with PECAM antibody to visualise vessels. (A–D and F–I) Representative images at × 10 magnification and × 20 (image inset). Quantification of vessel number in CAKI tumours (E) and PC3 (J). Overall P<0.0001 One-way ANOVA P<0.0001 **P<0.01, ***P<0.001 compared to control or VEGF165.
Discussion
VEGF has been generally considered in over 20 000 papers since 1990 as a pro-angiogenic tumour-enhancing endothelial-specific growth factor (Ferrara, 2002) and successful antiangiogenic agents have been directed at VEGF in cancer and eye disease (Gragoudas et al, 2004; Hurwitz et al, 2004). In 2002, we identified for the first time that an alternative splice site in the terminal exon 8 of the VEGF mRNA could be used to generate an alternative isoform (Bates et al, 2002), VEGF165b, which we subsequently showed to be one of a family of VEGF isoforms generated by C-terminal distal splice site selection, the VEGFxxxb family of isoforms (Perrin et al, 2005). However, while the conventional exon 8a containing isoforms predominate in the pathological angiogenic phenotype seen in tumours, proliferative retinopathy, arthritis etc (Ferrara, 2002), the exon 8b containing isoforms, which are anti-angiogenic in vivo are downregulated in a number of pathologies (Bates et al, 2002; Woolard et al, 2004; Cebe Suarez et al, 2006; Schumacher et al, 2007). Loss of the C-terminal domain (resulting in VEGF159) results in a loss of angiogenic activity of the VEGF molecule, but does not result in inhibition of angiogenesis (Cebe Suarez et al, 2006). The mechanism of action through which VEGFxxxb prevents tumour growth is not yet fully elucidated. However, it is clear from previous studies that VEGF165b is able to bind both VEGFR-1 (Cebe Suarez et al, 2006) and VEGFR-2 (Woolard et al, 2004), but initiates only weak signalling of the receptor to induce tyrosine phosphorylation (Woolard et al, 2004; Cebe Suarez et al, 2006), and is unable to induce a behavioural change in large vessel endothelial cells that mimic that shown by microvascular endothelial cells in vivo during angiogenesis (Woolard et al, 2004; Cebe Suarez et al, 2006). This study indicates for the first time that VEGF165b can exert its action by preventing tumour secreted endothelial growth factors (presumably VEGF165, or other VEGFxxx isoforms) from acting on microvascular endothelial cells.
Several studies have shown that VEGF expression in the malignant tissue and/or plasma correlates with aggressive disease (reviewed in Delongchamps et al, 2006) and VEGF mRNA and protein are upregulated in prostate carcinoma (Ferrer et al, 1997; Jackson et al, 1997). Our data indicate that there is a switch in VEGF expression allowing the pro-angiogenic VEGFxxx isoforms to dominate within malignant prostate and renal cell carcinoma, allowing the tumours to develop their own blood supply.
The current findings indicate that VEGF165b may have a therapeutic role in cancer treatment, by altering splicing of the VEGF gene to result in over-expression of VEGF165b at the expense of VEGF165. The latter mechanisms (control of splicing at the C-terminal end of the VEGF gene), is therefore one of intense interest, but unfortunately almost nothing has been published concerning the regulation of splicing of the VEGF gene. The vascular phenotype in both pathological and physiological angiogenesis may therefore depend on the balance of VEGF isoforms. We have speculated then that in addition to malignant change related to cell turnover/survival (which may also be determined by splicing (Venables, 2004) a second event occurs in which splicing control of many factors with pre- and antiangiogenic splice variants occur to allow the malignant disease to progress (Ladomery et al, 2006).
To conclude, these findings indicate that VEGF165b is able to inhibit growth of at least three different tumour types, and that the mechanism of inhibition is through inhibiting angiogenesis rather than a direct effect on tumour cell growth.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ (2002) VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is downregulated in renal cell carcinoma. Cancer Res 62: 4123–4131
Cebe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, Ballmer-Hofer K (2006) A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci 63: 2067–2077
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159
Delongchamps NB, Peyromaure M, Dinh-Xuan AT (2006) Role of vascular endothelial growth factor in prostate cancer. Urology 68: 244–248
Ferrara N (2002) Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol 29: 10–14
Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 25: 581–611
Ferrer FA, Miller LJ, Andrawis RI, Kurtzman SH, Albertsen PC, Laudone VP, Kreutzer DL (1997) Vascular endothelial growth factor (VEGF) expression in human prostate cancer: in situ and in vitro expression of VEGF by human prostate cancer cells. J Urol 157: 2329–2333
Gragoudas ES, Adamis AP, Cunningham Jr ET, Feinsod M, Guyer DR (2004) Pegaptanib for neovascular age-related macular degeneration. N Engl J Med 351: 2805–2816
Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW (1991) The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol 5: 1806–1814
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342
Jackson MW, Bentel JM, Tilley WD (1997) Vascular endothelial growth factor (VEGF) expression in prostate cancer and benign prostatic hyperplasia. J Urol 157: 2323–2328
Kusters B, Kats G, Roodink I, Verrijp K, Wesseling P, Ruiter DJ, de Waal RM, Leenders WP (2007) Micronodular transformation as a novel mechanism of VEGF-A-induced metastasis. Oncogene 26: 5808–5815
Ladomery MR, Harper SJ, Bates DO (2006) Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett 249 (2): 133–142
Mezquita P, Parghi SS, Brandvold KA, Ruddell A (2005) Myc regulates VEGF production in B cells by stimulating initiation of VEGF mRNA translation. Oncogene 24: 889–901
Miller-Kasprzak E, Jagodzinski PP (2008) 5-Aza-2-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed Pharmacother 62 (in press)
Perrin RM, Konopatskaya O, Qiu Y, Harper S, Bates DO, Churchill AJ (2005) Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia 48: 2422–2427
Pritchard-Jones RO, Dunn DB, Qiu Y, Varey AH, Orlando A, Rigby H, Harper SJ, Bates DO (2007) Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br J Cancer 97 (2): 223–230
Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS (1995) Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 55: 4575–4580
Schumacher VA, Jeruschke S, Eitner F, Becker JU, Pitschke G, Ince Y, Miner JH, Leuschner I, Engers R, Everding AS, Bulla M, Royer-Pokora B (2007) Impaired glomerular maturation and lack of VEGF165b in Denys-Drash syndrome. J Am Soc Nephrol 18: 719–729
Shweiki D, Itin A, Soffer D, Keshet E (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359: 843–845
Venables JP (2004) Aberrant and alternative splicing in cancer. Cancer Res 64: 7647–7654
Westphal JR, Van't Hullenaar R, Peek R, Willems RW, Crickard K, Crickard U, Askaa J, Clemmensen I, Ruiter DJ, De Waal RM (2000) Angiogenic balance in human melanoma: expression of VEGF, bFGF, IL-8, PDGF and angiostatin in relation to vascular density of xenografts in vivo. Int J Cancer 86: 768–776
Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R, Foster R, Digby-Bell J, Shields JD, Whittles CE, Mushens RE, Gillatt DA, Ziche M, Harper SJ, Bates DO (2004) VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 64: 7822–7835
Acknowledgements
We thank Leslie Sage for technical assistance. This work was supported by a Cancer Research UK Development Grant (A5047), the British Heart Foundation Grants (BB2000003 and BS06/005), National Kidney Research Fund Grant (R15/2/2003), the Prostate Research Campaign, Prostate Cancer Research Foundation, and the North Bristol Specific Cancer Projects Fund. The Ewing's Sarcoma experiments were carried out by Hui Guan and Eugenie Kleinerman at MD Anderson Cancer Center, Texas. The Mel57 experiments were carried out by William Leenders at the Radboud University, Nijmegen, The Netherlands.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rennel, E., Waine, E., Guan, H. et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br J Cancer 98, 1250–1257 (2008). https://doi.org/10.1038/sj.bjc.6604309
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604309
Keywords
This article is cited by
-
RNA splicing dysregulation and the hallmarks of cancer
Nature Reviews Cancer (2023)
-
How VEGF-A and its splice variants affect breast cancer development – clinical implications
Cellular Oncology (2022)
-
A drug-repositioning screen using splicing-sensitive fluorescent reporters identifies novel modulators of VEGF-A splicing with anti-angiogenic properties
Oncogenesis (2021)
-
VEGF-A165b levels are reduced in breast cancer patients at primary diagnosis but increase after completion of cancer treatment
Scientific Reports (2020)
-
In silico analysis of alternative splicing on drug-target gene interactions
Scientific Reports (2020)
