
Abstract
Capecitabine, a prodrug of 5-FU, has been reported to generate maximal tumour activity at tumour sites and/or to improve drug tolerability as compared with 5-FU infusion, and it has also been demonstrated to act synergistically with irinotecan against some solid cancers. A previous study concluded that dose-intensified biweekly capecitabine seems to be more effective at increasing both response rate and progression-free survival time than conventional dose and schedule of capecitabine in colon cancer. We conducted this study to ascertain the efficacy and toxicity of dose-intensified biweekly capecitabine and irinotecan combination chemotherapy in chemotherapy-naïve advanced or metastatic gastric cancer patients. Patients were treated with irinotecan 130 mg m−2 intravenously for 90 min on days 1 and 15. Capecitabine at 3500 mg m−2 day−1, divided into two sessions per day, was administered for seven consecutive days from days 1 and 15, and followed by a 7-day drug-free period, respectively. Fifty-five eligible patients were enrolled in this study from November 2003 to April 2006. There were 22 women and 33 men: median patient age was 54 years (range: 27–81). A total of 200 treatment cycles were administered at a median number of four per patient (range: 1–9). Intent-to-treatment analysis showed that one patient achieved complete response (1.8%), 23 partial response (41.8%), 15 stable disease (27.3%), 10 progressive disease (18.2%) and 6 were non-evaluable (10.9%). The overall response rate was 43.6% (95% confidence interval: 30.2–56.9). The common grade 3–4 toxicities were neutropenia in 12 (21.8%), nausea/vomiting in 3 (5.4%) and diarrhea in 4 (7.2%) patients. Median time to progression was 5 months (range: 0.5–11 months), median survival duration was 11 months (range: 0.5–45 months) and median response duration was 6 months (range: 0.5–9 months). Biweekly dose-intensified capecitabine and irinotecan combination chemotherapy was active for the treatment of advanced or metastatic gastric cancers with a tolerable safety profile.
Similar content being viewed by others

Main
Gastric cancer is the second most common cancer and one of the most common causes of cancer mortality in the world (Boring et al, 1994; Shin et al, 2005; Kamangar et al, 2006). Despite reducing gastric cancer incidence in the Occident, the incidence of cardiac and gastroesophageal junctional adenocarcinoma has increased, and the 5-year survival rate for advanced gastric cancer has not significantly changed (Brown and Devesa, 2002). The only curative treatment modality for gastric cancer is surgery. However, many patients present with advanced stage disease, and the relapse rate of those who are subjects for curative resection is high, at around 65% (Hartgrink et al, 2004). Thus, most gastric cancer patients are subjects for systemic chemotherapy. Current response rates to chemotherapy among advanced gastric cancer patients are between 10 and 20% for a single agent and between 30 and 50% for combination chemotherapy (Barone et al, 1998; Ajani et al, 2005; Moehler et al, 2005; Ohtsu et al, 2006; Nardi et al, 2007). However, during recent years, several new agents, for example, taxane, irinotecan and oxaliplatin, have been introduced for the treatment of gastric cancer (Louvet et al, 2002; Pozzo et al, 2004; Roth et al, 2004), and these drugs have shown promising response rates without sacrificing acceptable toxicity.
Irinotecan (Camptor®; Pfizer) is a semisynthetic plant alkaloid camptothecin, which inhibits DNA topoisomerase-I. SN-38 has been identified to be the important metabolite of irinotecan, and to inhibit the regulation of DNA during cell replication. In recent meta-analysis of chemotherapies used to treat advanced gastric cancer, a comparison between irinotecan-containing versus nonirinotecan-containing combinations (mainly 5-FU/cisplatin) showed a non-survival benefit in favour of irinotecan-containing regimens (HR=0.88) (Wagner et al, 2006).
Capecitabine (Xeloda®; Roche) is an oral fluoropyrimidine that selectively generates higher levels of 5-FU in cancer tissues than in normal tissues via the action of thymidine phosphorylase. Capecitabine monotherapy was proved to be active against advanced gastric cancer with the response rate of from 19.4 to 34% (Koizumi et al, 2003; Hong et al, 2004); moreover, median survival duration in these studies was comparable to other double or triple combination chemotherapies. Capecitabine has the advantages of convenient oral administration and of mimicking the effect of protracted intravenous (i.v.) 5-FU.
Irinotecan and capecitabine in combination could be possible to have a synergistic effect in previous pre-clinical and clinical studies (Guichard et al, 1998; Mullany et al, 1998; Azrak et al, 2004; Cao et al, 2005; Rea et al, 2005). Irinotecan reduces DNA synthesis, increases dTTP pools and inhibits dUMP synthesis, which are also associated with the antitumour activity of capecitabine (Mullany et al, 1998). Actually, irinotecan/capecitabine combination regimens have been used to treat several types of solid tumours (Borner et al, 2005; Han et al, 2005; Kim et al, 2005; Baek et al, 2006).
However, no optimum schedule has been identified for these two drugs. This is the first trial of high-dose regimen compared to usual capecitabine schedules and it is biweekly rather than triweekly against advanced gastric cancer. A preclinical study using human tumour xenografts indicated that tumour growth inhibition depends on the total dose of capecitabine administered and not on the dosing schedule (Van Cutsem et al, 2000), because short-term intermittent rather than prolonged or continuous administration may allow capecitabine dose intensities to be increased (and thus antitumour efficacies) without increasing toxicity via the incorporation of longer drug-free intervals. Actually, this dose-intensified biweekly administration seems to be more effective at increasing response rate and progression-free survival than conventional dose and schedule of capecitabine in colon cancer, without compromising its safety profile, despite the fact that capecitabine was administered with oxaliplatin (Thomas et al, 2001; Scheithauer et al, 2002, 2003). In addition, favourable results were reported for higher cumulative doses of oral capecitabine in advanced gastroesophageal cancer, although the administration schedule used differed from that used in the present study (Van Cutsem et al, 2000).
Here, we report the findings of a phase II study that was undertaken to evaluate the efficacy and toxicity of biweekly irinotecan and high-dose capecitabine combination chemotherapy for the treatment of advanced and/or metastatic gastric cancer in a first-line treatment setting.
Patients and methods
Between November 2003 and January 2006, 55 patients were enrolled in this trial. Written informed consent was obtained from all study subjects, and the study protocol was approved by our institutional review board. All patients had histologically proven gastric adenocarcinoma and locally unresectable or recurrent/metastatic disease. Only patients with measurable disease were accepted. The eligibility requirements included a performance status of ⩽2 and a life expectancy of at least 12 weeks. Preclinical laboratory parameters included an absolute neutrophil count of >1.5 × 103 l−1 and a platelet count of >100 × 103/ l−1. Patients were required to have normal cardiac, renal and hepatic functions. Contraindications to entry included any active infectious process, active heart disease or any concomitant second primary cancer. Patients who had undergone previous chemotherapy, except adjuvant chemotherapy completed within 6 months before enrollment, were also excluded.
Treatment
Irinotecan 130 mg m−2 was administrated i.v. for 90 min on days 1 and 15. Capecitabine 3500 mg m−2 day−1 b.i.d. was administered for seven consecutive days from days 1 and 15, which was followed by a 7-day drug-free interval, respectively.
If a patient developed neutropenia (an absolute neutrophil count of <1000) of >5 days duration or febrile neutropenia, irinotecan and capecitabine dosages were reduced by 25% during subsequent treatment cycles. Treatment was interrupted when a patient had a non-haematologic toxicity of >grade 2 during treatment, and restarted at a 25% reduced dosage after the toxicity had resolved to a level of <grade 2. If a patient had severe toxicity at the time of a scheduled retreatment, drug administration was postponed until the toxicity had resolved. If drug administration was delayed for more than 3 weeks, the patient was excluded from the study. When a patient had diarrhea, oral loperamide was immediately started at 2 mg, and subsequently 1 mg was given every 2 h until symptoms were relieved. Acute onset diarrhea that occurred within 24 h was considered as a cholinergic symptom and was prevented with atropine. This above cycle was repeated every 4 weeks until tumour progression or the development of treatment intolerance.
Evaluation
All patients were examined clinically before enrolment by laboratory evaluation, which included a complete blood cell count and electrolytes, blood urea nitrogen, creatinine, calcium, phosphorous, bilirubin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, total proteins and albumin. Upper gastrointestinal endoscopy, abdominal-computed tomography and other appropriate procedures were also performed. Complete blood cell counts were examined on days 1 and 14 of every cycle, and chest radiography, liver function tests, blood urea nitrogen, creatinine and electrolyte were examined on day 28 of every cycle. Examination of the evaluable parameters such as abdominal-computed tomography for evaluating of response was done after every two cycles of treatment. Response evaluation criteria in solid tumours were adopted to evaluate treatment response (Therasse et al, 2000). Toxicity was reported using a World Health Organization (WHO) Toxicity Criteria
Statistical analysis
Response rate was used as the primary end point in the present study, and a response rate of 40% was anticipated. A response rate of 20% was considered to be the minimum level for continuing the study. The α-level of the design used was 0.05 and its power was 0.9. The number of subjects was calculated using the Simon two-stage optimal Minimax design (Simon, 1989). A total of 45 evaluable patients were needed; 24 patients were planned for stage 1 and 21 for stage 2. If five or fewer responses had been observed during stage 1, then the trial would have been stopped. The proportion of patients who responded to treatment was used to estimate the true response rate with a 95% confidence interval (CI). Actuarial survival duration was determined using the Kaplan–Meier method. Response duration was calculated from the date of response confirmation to the date when disease progression was first observed. Survival duration was calculated from the first day of treatment until death or last follow-up. Time to disease progression was calculated from the first day of treatment to the date when disease progression was first observed.
Results
Patient's characteristics
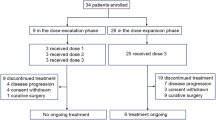
Fifty-five patients were enrolled in this trial. Patient characteristics are listed in Table 1. All patients were evaluable for toxicity, but only 49 patients (after excluding three patients who withdrew consent and three patients who failed to continue treatment due to adverse events after one cycle of chemotherapy) were evaluable. Median patient age was 54 years, and ranged from 27 to 81 years. Thirty-three patients were male and 22 were female. All patients had a good performance status of <grade 2. Twelve patients had a history of surgery (with curative intent for eight patients and with palliative intent for four), and only four patients had received adjuvant chemotherapy. Forty-three patients were treatment näive.
Response to chemotherapy
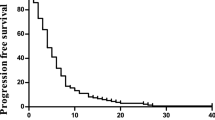
Fifty-five enrolled patients received 200 chemotherapy cycles; a median number of four per patient, which ranged from one to nine (Table 2). Six patients were non-evaluable; three patients refused further treatment after one cycle of chemotherapy and asked to be referred to other hospital, and treatment was discontinued in three patients due to toxicity after one cycle of chemotherapy. However, all six were included in the toxicity evaluation and in response rate calculations. By intention-to-treat analysis, one of the 55 patients achieved complete response (1.8%) and 23 showed partial response (41.8%). The overall objective response rate was 43.6% (95% CI: 30.2–56.9%) with a median response duration of 6 months (range: 0.5–9 months). Fifteen patients (27.3%) had stable disease and 10 patients (18.2%) had disease progression as a best response.
Median time to progression was 5 months (range: 0.5–11 months), and median survival duration was 11 months (range: 0.5–45 months).
Toxicity
Fifty-five patients were evaluable for toxicities (Table 3). The most commonly encountered haematologic grade 3 or 4 toxicity on irinotecan/capecitabine was neutropenia. Grades 3 and 4 neutropenia were experienced by eight (14.5%) and four patients (7.3%), respectively. Grades 3 and 4 anemia were also observed in five (9.1%) and one patient (1.8%), respectively. The most common non-haematologic toxicity was diarrhea. Grades 3 and 4 diarrhea both occurred in two patients (3.6%).
Fourteen patients required irinotecan and capecitabine dose modifications. Of these, the most common causes of dose reduction were prolonged grade 3 or 4 neutropenia in five patients. Other causes of dose reduction were grade 3 or 4 nausea/vomiting in one patient, grade 4 diarrhea in one patient and grade 3 fatigue in one patient. Another five patients had a capecitabine dose modification. The most common causes of these dose reductions were grade 2 or 3 hand/foot syndrome in three patients. Another two patients underwent capecitabine dose reduction due to investigator discretion for grade 3 fatigue in one patient and grade 3 hepatitis in the other. Three patients discontinued treatment due to toxicity after one cycle of chemotherapy; grade 3 ischemic colitis in one, bowel perforation in one and death from pneumonia in one. In total nine (4.5%) cycles were delayed. The most common cause of treatment delay was neutropenia in four patients; other causes were nausea/vomiting in three patients, fatigue in one and fever in one.
The mean dose intensities of irinotecan and capecitabine were 61.5 mg m−2 week−1 and 11 343 mg m−2 week−1, respectively, which were 94.7 and 92.6% of their respective intended doses (Figure 1).

Proportion of delivered dose over planned dose per cycle and per patient during respective treatment cycles.
Discussion
Pozzo et al (2004) compared irinotecan/cisplatin and irinotecan/5-FU/leucovorin with the aim of selecting a better regimen for a comparative phase III study with cisplatin/5-FU for the treatment of advanced gastric cancer, and the overall response rate of the irinotecan/5-FU/leucovorin arm (42%) was found to be superior to that of the irinotecan/cisplatin arm (32%). In the present study, we used capecitabine instead of 5-FU and leucovorin. Capecitabine is an oral fluoropyrimidine, and selectively produces higher concentrations of 5-FU in tumour tissues than in normal tissues. Hong et al (2004) studied capecitabine monotherapy for the treatment of advanced gastric cancer, and used capecitabine at 1250 mg m−2 twice daily (2500 mg m−2 day−1) for 14 days followed by 7 days of rest, for up to six cycles. Rates of overall response and stable disease were 34 and 30%, respectively, median time to progression was 3.2 months and median overall survival was 9.5 months. The most common toxicity was hand–foot syndrome, but grade 3 or 4 hand–foot syndrome was detected in only 9% of patients. Different capecitabine schedules have been examined for the treatment of advanced gastric cancer (Koizumi et al, 2003). In this earlier study, capecitabine as a single agent was administered at 828 mg m−2 b.i.d. for 3 weeks, followed by 1 week off, and a response rate of 19% (95% CI: 7.5–37.5%) was observed in gastric cancer. Median time to progression was 85 days and median survival was 247.5 days. No toxicity of ⩾grade 3 was reported, and the toxicity of capecitabine appeared to be correlated with the drug administration schedule.
The aim of this study was to determine the response rate and toxicities of combined dose-intensified biweekly capecitabine and irinotecan for the treatment of advanced gastric cancer. Capecitabine in the combination chemotherapy was reported to be at least as effective as 5-FU and leucovorin without additional toxicity in a REAL-2 study (Sumpter et al, 2005). Moreover, capecitabine has several advantages over i.v. 5-FU for clinical applications – the first of which is convenience. Capecitabine can be administered enterally without a portable infusion pump and central venous line, which results in high drug compliance and the maintenance of a constant 5-FU plasma level. The second reason concerns its safety, that is, capecitabine can be discontinued whenever severe toxicity develops, which prevents toxicity-induced deterioration. Our results demonstrate that dose-intensified biweekly capecitabine and irinotecan are active against advanced gastric cancer with a 43% overall response rate (95% CI: 30.2–56.9%) and a median response duration of 6 months. Moreover, the results achieved in this study could be favourably comparable with those of other studies that used an irinotecan-based combination regimen (Pozzo et al, 2004; Baek et al, 2006). Biweekly irinotecan and capecitabine (180 mg m−2, day 1 and 2000 mg m−2, days 1–9, respectively) have also been studied against gastroesophageal cancer (Burge et al, 2006), and an overall response rate of 32% was obtained. Because previous studies have produced improved results for dose-intensified biweekly capecitabine against colorectal cancer, in the present study, we adopted this dose and schedule for capecitabine instead of the conventional triweekly regimen against gastric cancer. The dose-intensified biweekly capecitabine arm showed a higher response rate (54.2 vs 42.2%) and a longer median progression-free survival time than the low-dose arm (10.5 vs 6.0 months) without an increase in toxicities (Van Cutsem et al, 2000; Scheithauer et al, 2003). In a study by Scheithauer et al, colorectal cancer patients received oxaliplatin 130 mg m−2 on day 1 plus capecitabine 2000 mg m−2 day−1 on days 1–14 every 3 weeks or oxaliplatin 85 mg m−2 on days 1 and 15 combined with capecitabine 3500 mg m−2 on days 1–7 and 15–21 every 4 weeks. However, no difference was found between the rates of haematologic and non-haematologic toxicities for the biweekly and triweekly regimens. Moreover, despite the use of a greater dose intensity of capecitabine, toxicities were tolerable for most patients, which were contrary to expectation (Kohne et al, 2005).
Clinical trials with novel drugs introduced to treat advanced gastric cancer have prolonged survival time. In particular, taxane- or oxaliplatin-based combination chemotherapies have shown high response rates from 38 to 54% and a median overall survival time of from 8.6 to 11.4 months against advanced gastric cancer (Louvet et al, 2002; De Vita et al, 2005; Lordick et al, 2005; Van Cutsem et al, 2006). Median survival duration in the present study was 11 months, which is the highest recorded to date by gastric cancer studies.
In the present study, neutropenia was the most common grade 3 or 4 haematologic toxicity, and was demonstrated in eight (14.5%) and four (7.3%) patients, respectively. However, all these patients recovered with supportive care based on, for example, the administration of granulocyte colony stimulating factor.
As was expected, in the present study, the most common non-haematologic toxicity was diarrhea, which occurred in 19 patients (34.5%). However, grade 3 or 4 diarrhea occurred in only two patients (3.6%) each. Moreover, this 7.2% rate of grade 3 or 4 diarrhea was less than that observed in other studies, which reported rates of 15–22% (Kim et al, 2005; Baek et al, 2006; Burge et al, 2006). The diarrhea and hand–foot syndrome were dose-limiting toxicities in phase I studies using capecitabine (Budman et al, 1998; Mackean et al, 1998). In the present study, nine patients (16.4%) experienced any grade of hand–foot syndrome and only three patients (5.5%) experienced grade 3 hand–foot syndrome. The rate of hand–foot syndrome in the present study was definitely lower than those of triweekly capecitabine studies, which have reported rates between 40 and 71% (Park et al, 2004; Baek et al, 2006). Despite a high mean dose intensity, the rates of diarrhea and hand–foot syndrome in the present study were lower than those of triweekly capecitabine studies (Hong et al, 2004; Park et al, 2004; Han et al, 2005; Baek et al, 2006). The capecitabine dose intensity used in the present study was 30% higher than that used by Baek et al (i.e., 11 343 mg m−2 week−1 in our study vs 8641 mg m−2 week−1 by Baek et al) and dose intensity was maintained during continued treatment cycles. In addition, the dose reduction rate in the present study (38.7%) was lower than that found by Baek et al study (57%) (Baek et al, 2006). Given higher dose intensities and uninterrupted treatment due to toxicity development, biweekly dose-intensified capecitabine and irinotecan combination chemotherapy appears to offer the possibility of longer response and survival duration.
In conclusion, dose-intensified biweekly capecitabine and irinotecan combination was found to be a moderately active regimen against advanced gastric cancer with manageable toxicity. The convenience of administering this regimen and the low rate of non-haematologic toxicities encountered suggest that it should be considered an option for the treatment of advanced gastric cancer. Phase III-randomized clinical trials are warranted to compare the efficacies and safety profiles of biweekly dose-intensified administration and conventional triweekly capecitabine.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajani JA, Fodor MB, Tjulandin SA, Moiseyenko VM, Chao Y, Cabral Filho S, Majlis A, Assadourian S, Van Cutsem E (2005) Phase II multi-institutional randomized trial of docetaxel plus cisplatin with or without fluorouracil in patients with untreated, advanced gastric, or gastroesophageal adenocarcinoma. J Clin Oncol 23: 5660–5667
Azrak RG, Cao S, Slocum HK, Toth K, Durrani FA, Yin MB, Pendyala L, Zhang W, McLeod HL, Rustum YM (2004) Therapeutic synergy between irinotecan and 5-fluorouracil against human tumor xenografts. Clin Cancer Res 10: 1121–1129
Baek JH, Kim JG, Jeon SB, Chae YS, Kim DH, Sohn SK, Lee KB, Choi YJ, Shin HJ, Chung JS, Cho GJ, Jung HY, Yu W (2006) Phase II study of capecitabine and irinotecan combination chemotherapy in patients with advanced gastric cancer. Br J Cancer 94: 1407–1411
Barone C, Corsi DC, Pozzo C, Cassano A, Fontana T, Noviello MR, Landriscina M, Colloca G, Astone A (1998) Treatment of patients with advanced gastric carcinoma with a 5-fluorouracil-based or a cisplatin-based regimen: two parallel randomized phase II studies. Cancer 82: 1460–1467
Boring CC, Squires TS, Tong T, Montgomery S (1994) Cancer statistics, 1994. CA Cancer J Clin 44: 7–26
Borner MM, Bernhard J, Dietrich D, Popescu R, Wernli M, Saletti P, Rauch D, Herrmann R, Koeberle D, Honegger H, Brauchli P, Lanz D, Roth AD (2005) A randomized phase II trial of capecitabine and two different schedules of irinotecan in first-line treatment of metastatic colorectal cancer: efficacy, quality-of-life and toxicity. Ann Oncol 16: 282–288
Brown LM, Devesa SS (2002) Epidemiologic trends in esophageal and gastric cancer in the United States. Surg Oncol Clin N Am 11: 235–256
Budman DR, Meropol NJ, Reigner B, Creaven PJ, Lichtman SM, Berghorn E, Behr J, Gordon RJ, Osterwalder B, Griffin T (1998) Preliminary studies of a novel oral fluoropyrimidine carbamate: capecitabine. J Clin Oncol 16: 1795–1802
Burge ME, Smith D, Topham C, Jackson DP, Anthoney DA, Halstead F, Seymour MT (2006) A phase I and II study of 2-weekly irinotecan with capecitabine in advanced gastroesophageal adenocarcinoma. Br J Cancer 94: 1281–1286
Cao S, Durrani FA, Rustum YM (2005) Synergistic antitumor activity of capecitabine in combination with irinotecan. Clin Colorectal Cancer 4: 336–343
De Vita F, Orditura M, Matano E, Bianco R, Carlomagno C, Infusino S, Damiano V, Simeone E, Diadema MR, Lieto E, Castellano P, Pepe S, De Placido S, Galizia G, Di Martino N, Ciardiello F, Catalano G, Bianco AR (2005) A phase II study of biweekly oxaliplatin plus infusional 5-fluorouracil and folinic acid (FOLFOX-4) as first-line treatment of advanced gastric cancer patients. Br J Cancer 92: 1644–1649
Guichard S, Hennebelle I, Bugat R, Canal P (1998) Cellular interactions of 5-fluorouracil and the camptothecin analogue CPT-11 (irinotecan) in a human colorectal carcinoma cell line. Biochem Pharmacol 55: 667–676
Han JY, Lee DH, Lee SY, Park CG, Kim HY, Lee HG, Lee JJ, Kim HT, Lee JS (2005) Phase II study of weekly irinotecan plus capecitabine for chemotherapy-naive patients with advanced nonsmall cell lung carcinoma. Cancer 104: 2759–2765
Hartgrink HH, van de Velde CJ, Putter H, Bonenkamp JJ, Klein Kranenbarg E, Songun I, Welvaart K, van Krieken JH, Meijer S, Plukker JT, van Elk PJ, Obertop H, Gouma DJ, van Lanschot JJ, Taat CW, de Graaf PW, von Meyenfeldt MF, Tilanus H, Sasako M (2004) Extended lymph node dissection for gastric cancer: who may benefit? Final results of the randomized Dutch gastric cancer group trial. J Clin Oncol 22: 2069–2077
Hong YS, Song SY, Lee SI, Chung HC, Choi SH, Noh SH, Park JN, Han JY, Kang JH, Lee KS, Cho JY (2004) A phase II trial of capecitabine in previously untreated patients with advanced and/or metastatic gastric cancer. Ann Oncol 15: 1344–1347
Kamangar F, Dores GM, Anderson WF (2006) Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol 24: 2137–2150
Kim TW, Kang WK, Chang HM, Park JO, Ryoo BY, Ahn JS, Zang DY, Lee KH, Kang YK, Kim SR, Kim HK (2005) Multicenter phase II study of oral capecitabine plus irinotecan as first-line chemotherapy in advanced colorectal cancer: a Korean Cancer Study Group trial. Acta Oncol 44: 230–235
Kohne C, de Greve J, Bokemeyer C, Lang I, Vergauwe P, Braumann D, Debois M, Meulemans B, Therasse P, van Cutsem E (2005) Capecitabine plus irinotecan versus 5-FU/FA/irinotecan +/− celecoxib in first line treatment of metastatic colorectal cancer. Safety results of the prospective multicenter EORTC phase III study 40015. J Clin Oncol, 2005 ASCO Annual Meeting Proceedings 23: 16s (abstract 3525)
Koizumi W, Saigenji K, Ujiie S, Terashima M, Sakata Y, Taguchi T (2003) A pilot phase II study of capecitabine in advanced or recurrent gastric cancer. Oncology 64: 232–236
Lordick F, Lorenzen S, Stollfuss J, Vehling-Kaiser U, Kullmann F, Hentrich M, Zumschlinge R, Dietzfelbinger H, Thoedtmann J, Hennig M, Seroneit T, Bredenkamp R, Duyster J, Peschel C (2005) Phase II study of weekly oxaliplatin plus infusional fluorouracil and folinic acid (FUFOX regimen) as first-line treatment in metastatic gastric cancer. Br J Cancer 93: 190–194
Louvet C, Andre T, Tigaud JM, Gamelin E, Douillard JY, Brunet R, Francois E, Jacob JH, Levoir D, Taamma A, Rougier P, Cvitkovic E, de Gramont A (2002) Phase II study of oxaliplatin, fluorouracil, and folinic acid in locally advanced or metastatic gastric cancer patients. J Clin Oncol 20: 4543–4548
Mackean M, Planting A, Twelves C, Schellens J, Allman D, Osterwalder B, Reigner B, Griffin T, Kaye S, Verweij J (1998) Phase I and pharmacologic study of intermittent twice-daily oral therapy with capecitabine in patients with advanced and/or metastatic cancer. J Clin Oncol 16: 2977–2985
Moehler M, Eimermacher A, Siebler J, Hohler T, Wein A, Menges M, Flieger D, Junginger T, Geer T, Gracien E, Galle PR, Heike M (2005) Randomised phase II evaluation of irinotecan plus high-dose 5-fluorouracil and leucovorin (ILF) vs 5-fluorouracil, leucovorin, and etoposide (ELF) in untreated metastatic gastric cancer. Br J Cancer 92: 2122–2128
Mullany S, Svingen PA, Kaufmann SH, Erlichman C (1998) Effect of adding the topoisomerase I poison 7-ethyl-10-hydroxycamptothecin (SN-38) to 5-fluorouracil and folinic acid in HCT-8 cells: elevated dTTP pools and enhanced cytotoxicity. Cancer Chemother Pharmacol 42: 391–399
Nardi M, Azzarello D, Maisano R, Del Medico P, Giannicola R, Raffaele M, Zavettieri M, Costarella S, Falzea A (2007) FOLFOX-4 regimen as first-line chemotherapy in elderly patients with advanced gastric cancer: a safety study. J Chemother 19: 85–89
Ohtsu A, Yoshida S, Saijo N (2006) Disparities in gastric cancer chemotherapy between the East and West. J Clin Oncol 24: 2188–2196
Park SH, Bang SM, Cho EK, Baek JH, Oh JH, Im SA, Park YS, Shin DB, Lee JH (2004) First-line chemotherapy with irinotecan plus capecitabine for advanced colorectal cancer. Oncology 66: 353–357
Pozzo C, Barone C, Szanto J, Padi E, Peschel C, Bukki J, Gorbunova V, Valvere V, Zaluski J, Biakhov M, Zuber E, Jacques C, Bugat R (2004) Irinotecan in combination with 5-fluorouracil and folinic acid or with cisplatin in patients with advanced gastric or esophageal-gastric junction adenocarcinoma: results of a randomized phase II study. Ann Oncol 15: 1773–1781
Rea DW, Nortier JW, Ten Bokkel Huinink WW, Falk S, Richel DJ, Maughan T, Groenewegen G, Smit JM, Steven N, Bakker JM, Semiond D, Kerr DJ, Punt CJ (2005) A phase I/II and pharmacokinetic study of irinotecan in combination with capecitabine as first-line therapy for advanced colorectal cancer. Ann Oncol 16: 1123–1132
Roth AD, Maibach R, Falk S, Stupp R, Saletti P, Kãberle D, Borner MM, Honegger H-P, Leslie M, Fazio N (2004) Docetaxel-cisplatin-5FU (TCF) versus docetaxel-cisplatin (TC) versus epirubicin-cisplatin-5FU (ECF) as systemic treatment for advanced gastric carcinoma (AGC): a randomized phase II trial of the Swiss Group for Clinical Cancer Research (SAKK). J Clin Oncol 22: 318S
Scheithauer W, Kornek GV, Raderer M, Schull B, Schmid K, Kovats E, Schneeweiss B, Lang F, Lenauer A, Depisch D (2003) Randomized multicenter phase II trial of two different schedules of capecitabine plus oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 21: 1307–1312
Scheithauer W, Kornek GV, Raderer M, Schull B, Schmid K, Langle F, Huber H (2002) Intermittent weekly high-dose capecitabine in combination with oxaliplatin: a phase I/II study in first-line treatment of patients with advanced colorectal cancer. Ann Oncol 13: 1583–1589
Shin H-R, Won Y-J, Jung K-W, Kong H-J, Yim S-H, Lee J-K, Noh H-I, Lee J-K, Pisani P, Park J-G, Members of the Regional Cancer Registries (2005) Nationwide cancer incidence in Korea, 1999–2001: first result using the National Cancer Incidence Database. Cancer Res Treat 37: 325–331
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10: 1–10
Sumpter K, Harper-Wynne C, Cunningham D, Rao S, Tebbutt N, Norman AR, Ward C, Iveson T, Nicolson M, Hickish T, Hill M, Oates J (2005) Report of two protocol planned interim analyses in a randomised multicentre phase III study comparing capecitabine with fluorouracil and oxaliplatin with cisplatin in patients with advanced oesophagogastric cancer receiving ECF. Br J Cancer 92: 1976–1983
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216
Thomas R, Quinn M, Wilson R, Floeter MK, Lehky T, Wasif Saif J, Hamilton M, Monahan B, Grochow L, Harold N, Schuler B, Allegra C, Cliatt J, Grem J (2001) A Phase I Trial of Capecitabine (CAPE) & Oxaliplatin (OHP). Proc Am Soc Clin Oncol 20: 133a (abstract 530)
Van Cutsem E, Findlay M, Osterwalder B, Kocha W, Dalley D, Pazdur R, Cassidy J, Dirix L, Twelves C, Allman D, Seitz JF, Scholmerich J, Burger HU, Verweij J (2000) Capecitabine, an oral fluoropyrimidine carbamate with substantial activity in advanced colorectal cancer: results of a randomized phase II study. J Clin Oncol 18: 1337–1345
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, Risse ML, Ajani JA (2006) Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24: 4991–4997
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE (2006) Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol 24: 2903–2909
Acknowledgements
This study was supported in part by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A040151 and A010250).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Oh, S., Sur, H., Sung, H. et al. A phase II study of biweekly dose-intensified oral capecitabine plus irinotecan (bXELIRI) for patients with advanced or metastatic gastric cancer. Br J Cancer 96, 1514–1519 (2007). https://doi.org/10.1038/sj.bjc.6603752
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603752
Keywords
This article is cited by
-
Role of low dose capecitabine combined to irinotecan in advanced and metastatic gastric cancer
Medical Oncology (2010)
-
A phase II trial evaluating capecitabine and irinotecan as second line treatment in patients with oesophago-gastric cancer who have progressed on, or within 3 months of platinum-based chemotherapy
Cancer Chemotherapy and Pharmacology (2009)
-
ERCC1 mRNA levels and survival of advanced gastric cancer patients treated with a modified FOLFOX regimen
British Journal of Cancer (2008)
