
Abstract
The selection of candidates for BRCA germline mutation testing is an important clinical issue yet it remains a significant challenge. A number of risk prediction models have been developed to assist in pretest counselling. We have evaluated the performance and the inter-rater reliability of four of these models (BRCAPRO, Manchester, Penn and the Myriad-Frank). The four risk assessment models were applied to 380 pedigrees of families who had undergone BRCA1/2 mutation analysis. Sensitivity, specificity, positive and negative predictive values, likelihood ratios and area under the receiver operator characteristic (ROC) curve were calculated for each model. Using a greater than 10% probability threshold, the likelihood that a BRCA test result was positive in a mutation carrier compared to the likelihood that the same result would be expected in an individual without a BRCA mutation was 2.10 (95% confidence interval (CI) 1.66–2.67) for Penn, 1.74 (95% CI 1.48–2.04) for Myriad, 1.35 (95% CI 1.19–1.53) for Manchester and 1.68 (95% CI 1.39–2.03) for BRCAPRO. Application of these models, therefore, did not rule in BRCA mutation carrier status. Similar trends were observed for separate BRCA1/2 performance measures except BRCA2 assessment in the Penn model where the positive likelihood ratio was 5.93. The area under the ROC curve for each model was close to 0.75. In conclusion, the four models had very little impact on the pre-test probability of disease; there were significant clinical barriers to using some models and risk estimates varied between experts. Use of models for predicting BRCA mutation status is not currently justified for populations such as that evaluated in the current study.
Similar content being viewed by others


Main
Germline mutations in BRCA1 and BRCA2 confer an estimated 65 and 45% cumulative lifetime risk of developing breast cancer, and an ovarian cancer risk of 39 and 11%, respectively (Antoniou et al, 2003). Identification of these individuals before they present with cancer is important since prophylactic surgery can reduce morbidity and mortality in these individuals (Struewing et al, 1995; Rebbeck et al, 1999; Hartmann et al, 1999; Hartmann et al, 2001; Meijers-Heijboer et al, 2001; Kauff et al, 2002; Rebbeck et al, 2004). Unfortunately, genetic testing is expensive, family history of breast cancer is common and BRCA mutations are rare (Ford et al, 1994; Peto et al, 1999; Antoniou et al, 2002). A triage tool to identify those families most likely to benefit from germline testing would, therefore, be very useful. Like most countries (summarized by Nelson et al, 2005), Australia has developed guidelines (NBCC Genetics Working Group, 2000) for the referral of individuals to a family cancer clinic for counselling and further evaluation. Under these guidelines, full BRCA1/2 mutation screening is usually restricted to individuals affected by breast or epithelial ovarian cancer, and whose family history as a whole satisfies a ‘potentially high-risk category’ (NBCC Genetics Working Group, 2000; Nelson et al, 2005). The prevalence of germline BRCA mutations in such high-risk families is estimated at 3.4–15.5% (Frank et al, 2002; Huang et al, 2002; Antoniou et al, 2003; Myriad, 2004; Nelson et al, 2005). These low figures have led to the development of models that can more accurately assess the pre-test probability of identifying a BRCA1/2 germline mutation (Couch et al, 1997; Parmigiani et al, 1998; Berry et al, 2002; Frank et al, 2002; Evans et al, 2004). Mendelian models like BRCAPRO (Parmigiani et al, 1998; Berry et al, 2002) evaluate the probability that an individual is a gene mutation carrier, while other models such as Penn (Couch et al, 1997), Manchester (Evans et al, 2004) and Frank-Myriad (Frank et al, 2002) determine the likelihood of identifying a mutation on the basis of known family history. Although the performance of some of these models has been previously examined, it is perhaps telling that no single model has been universally adopted. Previous model validation studies have considered only a subset of the available models, have not compared the results with germline testing, and have not considered the barriers to the use of models in clinical practice (Berry et al, 2002; Euhus et al, 2002; de la Hoya et al, 2003; Marroni et al, 2004; James et al, 2006; Barcenas et al, 2006).
This study sought to compare the ability of four models (BRCAPRO, Manchester, Penn and the Myriad-Frank) to determine the likelihood of finding a BRCA gene mutation in at-risk individuals using a large group of patients who had undergone germline testing.
Methods
Patient cohort
Family cancer clinics at St Vincent’s and Westmead Hospitals, Sydney, provided pedigrees of families who had undergone BRCA1/2 mutation analysis in the period 1998–2004. BRCA1/2 testing was performed using the DNA of an affected individual, usually the youngest living available affected person, or an obligate gene mutation carrier. For the purposes of the study, this individual was defined as the proband. Families in which no affected individuals were available for BRCA1/2 testing were not included in the study. To be eligible for germline testing, at least one unaffected family member must have had a life time risk of breast cancer of 1 : 4 or greater as defined by the Australian National Breast Cancer (NBCC) guidelines (NBCC Genetics Working Group, 2000). This included individuals with at least two first- or second-degree relatives on one side of the family diagnosed with breast or ovarian cancer, together with additional features on the same side of the family. These features included an additional relative with breast or ovarian cancer; breast cancer diagnosed before the age of 40 years, ovarian cancer before 50 years, bilateral breast cancer, breast and ovarian cancer in the same woman, Jewish ancestry or breast cancer in a male relative.
Each pedigree was de-identified, with only the sex, birthday, date of death, type of cancer and age at diagnosis retained for the analysis. Not all cancers could be verified because of the requirement for client consent in Australia. Pedigrees were viewed with Progeny software v5.4.05 (Progeny Software, LLC, South Bend, IN, USA). Furthermore, families of Ashkenazi Jewish ancestry were not included in this study. This study was approved by the St Vincents Hospital Human Research Ethics Committee.
Use of models to identify mutation carriers
Each of the four risk assessment models (Manchester (Evans et al, 2004; Evans et al, 2005), Myriad (Frank et al, 2002), Penn (Couch et al, 1997) and BRCAPRO (Parmigiani et al, 1998; Berry et al, 2002)) were applied to each pedigree by two investigators (HK and RWi). All models except Myriad allow calculations for combined BRCA mutation status, as well as BRCA1 and BRCA2 independently. Risk assessments were based on the proband, and maternal or paternal inheritance was assessed using the Progeny program (Progeny Software, LLC, South Bend, IN, USA). If the potential carrier side was not obvious, the assessments were conducted for both paternal and maternal relatives and the highest score was included in the analysis. As data on some pedigrees was ambiguous or incomplete, prespecified rules were used for the entry of information into each model (Table 1). Determination of mutation likelihood using the Manchester model was performed by assigning a score on the basis of malignancy type and age of diagnosis (Evans et al, 2004). Scores obtained do not equate directly to probabilities; however, the authors suggest that a cutoff score of 10 points for BRCA1 and BRCA2 separately, or a combined score of 15 points correspond to an approximate mutation probability of 10% (Evans et al, 2004; Evans et al, 2005). The Myriad model was applied using on-line tables (Myriad, 2004) and an on-line questionnaire was used to obtain independent probabilities for BRCA1 and BRCA2 mutations using the Penn model (University of Pennsylvania Abramson Cancer Center, 2005). The latter model requires the presence of breast cancer in the family lineage and thus six families with only ovarian cancer could not be analysed. The overall chance of a mutation in either BRCA1 or BRCA2 was calculated as the probability of mutation in BRCA1 plus the probability of mutation in BRCA2 assuming there was no mutation in BRCA1. For the BRCAPRO model, carrier probabilities were calculated by entering information on the proband's first- and second-degree relatives into CancerGene software (CaGene version 3.3, supplied by Assistant Professor D Euhus, UT Southwestern Medical Center at Dallas, Texas, USA).
Mutation screening for BRCA1 and BRCA2
The extent to which BRCA1/2 were screened reflects the availability of testing methodologies over the study period. Full DNA sequence analysis of BRCA1/2 together with a screen for the BRCA1 exon 13 duplication (common in families of British descent) (Puget et al, 1999) was used as the minimum testing regime in 89 families. Mutation analysis on the remaining 291 families was performed by a screening strategy that involved the use of a number of different techniques. The protein truncation test (PTT) was performed for exon 11 of BRCA1 and exons 10, 11 and 27 of BRCA2. This test covers 65% of the coding region for each gene and identifies the most common types of pathogenic mutation in BRCA1/2 (Garvin, 1998). Sequence analysis or heteroduplex analysis of exons 2 and 20 of BRCA1 using either polyacrylamide gel electrophoresis or denaturing high-performance liquid chromatography were used to detect nonsense, frameshift and missense mutations within these exons. All samples were screened for the BRCA1 duplication exon 13 mutation and a subset of 125 families from the entire cohort were also screened for rare large genomic rearrangements in BRCA1 using multiplex ligation-dependent probe amplification (MLPA) (Woodward et al, 2005).
BRCA1/2 sequence variants of uncertain clinical significance were identified in nine cases and these families were excluded from the analysis; however, families with nondeleterious, common polymorphisms were included in the no-mutation group.
Statistical methods
All generated scores were analysed using SPSS statistical software V13.0 (SPSS Inc., Chicago, IL, USA). Sensitivity, specificity, positive and negative predictive values, and positive and negative likelihood ratios were calculated for each risk model at the 10% threshold. For the Manchester model, this threshold corresponded with a cutoff score of 10 points for BRCA1 and BRCA2 separately, and a score of 15 points for the combined model. The positive likelihood ratio of a model was calculated as the proportion of individuals with a BRCA1/2 mutation who have a model probability 10% or greater divided by the proportion of individuals without a mutation who have a model probability >10%. Similarly, the negative likelihood ratio was calculated as the ratio of the proportion of individuals with a BRCA1/2 mutation who have a model probability <10% to the proportion of individuals without a mutation who have a model probability <10%. Receiver operator characteristics (ROC) curves were constructed by plotting the sensitivity (or true positive fraction) against 1 minus specificity (or false positive fraction) for all possible values of the mutation probability. The area under the ROC curve (C-statistic) was calculated as a measure of the accuracy of the model for discriminating between mutation carriers and those without a BRCA mutation. It represents the fraction of all probands with identified family mutations that have a detection probability higher than a proband with no mutation identified in the family. The κ scores were used to assess the measure of agreement between 100 independent risk calculations from 100 pedigrees chosen at random. In this analysis, a score of 0–0.2 describes slight, 0.2–0.4 fair, 0.4–0.6 moderate, 0.6–0.8 substantial and over 0.8 almost perfect agreement (Koch et al, 1977).
Results
Proportion of positive BRCA tests varied according to carrier probability
The study cohort included pedigrees from 380 families, of whom 52 (13.7%) carried deleterious mutations in either BRCA1 (34 subjects) or BRCA2 (18 subjects). The mutation frequency was higher (22.5%) in individuals subjected to complete DNA sequencing of BRCA1/2 (20 of 89 individuals) compared with those in whom sequencing had not been performed (11%, 32 of 291 subjects).
A total of 45 different mutations were recorded among the BRCA mutation positive families, whereas duplication of BRCA1 exon 13 occurred in a further five families. The mean carrier probabilities for mutation-positive individuals were 53% using BRCAPRO, 26% for Myriad and 32% using the Penn model. The probabilities for mutation-negative individuals were 24, 14, and 12%, respectively. The mean score for mutation-positive individuals using the Manchester model was 37; the mean score for mutation-negative individuals was 21. All models showed a relationship between the carrier probability and the proportion of positive tests (Table 2).
Discrimination of BRCA mutation carriers from noncarriers
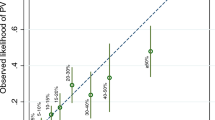
Table 3 shows the sensitivity, specificity, positive and negative predictive values for each risk model at the 10% threshold. The negative predictive value of all four models was remarkably consistent (range=0.93–0.96) in that only 4–6% of families assigned a mutation probability of 10% or less actually carried a mutation in BRCA1 or BRCA2 (Table 3). Given the relatively low prevalence of BRCA mutations in the study population, it is not surprising that the positive predictive value at the 10% threshold was low (range=0.18–0.25). The predictive value improved as the threshold probability was increased; however, 58% of probands assigned an 80% or greater mutation probability using BRCAPRO did not have a mutation detected (Table 2). All probands with a score of 80 or more according to the Manchester model carried a mutation. The likelihood that the model probability was >10% in a mutation carrier was 2.10 (95% CI 1.66–2.67) for Penn, 1.74 (95%CI 1.48–2.04) for Myriad, 1.35 (95% CI 1.19–1.53) for Manchester and 1.68 (95% CI 1.39–2.03) for BRCAPRO. Application of these models, therefore, does not rule in BRCA mutation carrier status. The negative likelihood ratios were consistently greater than 0.1 (BRCAPRO 0.43, Manchester 0.34, Myriad 0.30 and Penn 0.46), which indicated that the models are also unable to rule out mutation carrier status. The post-test probability for mutation carrier status (calculated by multiplying pre-test odds of disease by the positive or negative likelihood ratio) is shown in Figure 1. It is apparent that the estimated pre-test probability of a mutation is not significantly altered by calculating the mutation risk estimate using each of the models. For example, a prediction of a germline mutation for the Penn model would change an individual's pre-test mutation probability from 40 to 58%, whereas using the Manchester model it would increase from 40 to 47%.

Post-test probability for mutation carrier status is shown for each model using the positive and negative likelihood ratios.
The accuracy of the four models was also compared by examining the areas under the ROC curve (Figure 2 and Table 4). A model that correlates perfectly with BRCA mutation status would have an area under the curve of 1.0 while an area of 0.5 indicates that the model has no discriminatory value. Using the scores for detecting either a BRCA1 or BRCA2 mutation, the area under the curve for each model was close to 0.75 (Table 4). By this measure, the models were equal in their ability to discriminate between mutation carrying and noncarrying probands.

Receiver operator characteristics (ROC) curves for models comprising BRCA1, BRCA2 and the combination. Diagonal segments are produced by ties.
To evaluate the possibility that the mutation-testing strategy influences the performance of the models, we stratified the data according to type of gene sequencing results. Although the frequency of mutation carriers was higher in the group tested by BRCA1/2 sequencing, the models were found to perform similarly for those with and without complete DNA sequencing results. While positive predictive values of models tended to be higher for those with complete DNA sequencing, there were no significant differences in mean model scores or probabilities, sensitivity, specificity, predictive values, likelihood ratios and C-statistics (data not presented).
Differentiating mutations in BRCA1 and BRCA2
The Manchester, BRCAPRO and Penn models allow separate calculations of the probability of BRCA1 and BRCA2 mutations (Tables 3 and 5). The positive likelihood ratios for BRCA1 mutation carriers were 2.74 (95% CI 1.94–3.88) for Penn, 1.87 (95% CI 1.59–2.21) for Manchester and 2.15 (95% CI 1.72–2.67) for BRCAPRO. The negative likelihood ratios were 0.52, 0.22 and 0.36, respectively. The Penn model could potentially be used to rule in BRCA2 mutations, in that the positive likelihood ratio was 5.93 (95% CI 2.72–12.94) although the negative likelihood ratio was unacceptably high at 0.71 (95% CI 0.51–0.98). The Manchester and BRCAPRO had likelihood ratios around 1, indicating that these models provided no additional information in determining BRCA2 mutation carrier status.
A comparison of the respective ROC curves shows that the Manchester model, BRCAPRO and Penn performed similarly for BRCA1, but that BRCAPRO was the least accurate model for BRCA2 identification (Figure 2, Table 4). The proportions of BRCA1 and 2 mutation carriers at each threshold for the Manchester, BRCAPRO and Penn models are shown in Table 5.
Validation of risk calculation estimates
Given the inherent ambiguities in interpreting pedigrees, two of the investigators (HK and RWi) specified rules for dealing with various clinical scenarios (see methods and Table 1). At the completion of the study, a κ score of mutation-risk estimates using the models was determined for 100 randomly selected pedigrees (25 cases for each risk model). Overall, the κ score was 0.82 reflecting excellent agreement between observers when calculating the mutation risk for each proband. The measure of agreement differed between models in that perfect agreement was noted for Penn (κ=1.0) and Manchester (κ=0.932), whereas only substantial agreement was found for Myriad (κ=0.714) and for BRCAPRO (κ=0.60). The areas of disagreement in applying the BRCAPRO model were related to clinical judgment on choice of proband, estimation of age of relatives, and inclusion of maternal and paternal relatives.
Discussion
In this study we used the family histories of a large tested cohort, some with known BRCA germline mutations, to evaluate the clinical effectiveness of four risk prediction models for BRCA mutations. Estimation of pre-test mutation probability is an important clinical issue, particularly in view of the expense, legal implications and other difficulties associated with ambiguous germline BRCA testing results. Germline testing is currently recommended solely on the basis of clinical suspicion, yet even within this group of patients few will harbour a mutation (13% in the current study). While prediction models (Euhus, 2001; Domchek et al, 2003) offer an opportunity to improve clinical decision-making, to be effective in clinical practice they must be practical as well as accurate. Our study highlighted certain aspects of the models, which are likely to detract from their effective use in family cancer clinics. The Manchester model (Evans et al, 2004) and the Myriad mutation prevalence tables (Myriad, 2004) did not require computer access and were extremely rapid methods of assessing risk. Like most empiric models, the risk estimates relied on the ‘number of cases per family’, yet the definition of a family can be highly subjective. For instance, large pedigrees with many elderly female subjects with breast cancer generated a cumulative score above 10% using the Manchester model. On the other hand, the Myriad tables only allowed inclusion of a maximum of three members of the family, including the patient. Other disconcerting features of the Myriad model were that breast cancers diagnosed above 50 years were ignored, whereas for those diagnosed before 50 years there was no stratification according to the age of diagnosis. Further deficiencies included the equal weighting given to male and female breast cancers and the inability to input bilateral breast cancer or other tumours associated with BRCA1/2 mutation, namely prostate and pancreatic cancers (Lynch et al, 1999; Tulinius et al, 2002; Liede et al, 2004).
Both the Penn model and BRCAPRO required computer access. In the case of BRCAPRO, the time taken to enter family trees was a major impediment to routine use. BRCAPRO only incorporates first- and second-degree relatives and therefore cousins of the proband who are affected with cancer will not be used to generate a probability score unless the counselor changes the proband. This scenario was in part responsible for the low κ scores associated with the use of BRCAPRO. The on-line Penn model was a very rapid and efficient mode of calculating not only the individual but also the family’s probability of carrying a BRCA1/2 mutation. The model also provided question prompts with helpful explanatory comments; this feature significantly reduced the chance of disagreement between the independent risk assessors. Unfortunately, the Penn model also restricted questions to three generations, and did not include ovarian cancer only families or mother–daughter ovarian–breast cancer inheritance patterns.
In comparing the accuracy of the models, we found that they had an equal ability to discriminate between mutation carrying and noncarrying probands (C-statistic 0.743–0.759 for combined analysis using the ROC curves) when considering the entire range of possible test thresholds. These results are highly comparable to those of an Italian study of BRCA mutation carriers where risk was assessed using the Myriad tables, BRCAPRO and an old version of the Penn model (Marroni et al, 2004). The initial report of the Manchester model claimed superiority over BRCAPRO on the basis of a larger area under the ROC curve (0.772 vs 0.596) (Evans et al, 2004). We were unable to confirm these findings and note that the area under the curve reported by Evans et al for BRCAPRO was considerably lower than that found in the current study as well as a number of other studies (Euhus et al, 2002; Marroni et al, 2004). The source of these discrepancies may relate to the limitations of comparing a scoring system (Manchester) with probability calculations (BRCAPRO).
Although the area under the ROC curves was comparable between models, this does not mean that they are equally accurate in predicting germline mutation status at the well-accepted 10% probability threshold. Examination of the likelihood ratios allows such a comparison and also provides an assessment of the value of the models in relation to other tools. Penn was the most discriminatory model, in that individuals with BRCA mutation were twice as likely to have a positive prediction as those without a germline mutation. In contrast, the Manchester model had a ratio closer to unity indicating that it is unhelpful in clinical decision-making. In other areas of medicine, likelihood ratios greater than 5 are generally a prerequisite for the adoption of a clinical test or procedure. By way of comparison, the positive likelihood ratio for mammography is 14 (Kerlikowske et al, 1995), for bedside cardiac-specific troponin T 6.3 (Antman et al, 1995) and for ultrasonography for deep venous thrombosis 47.8 (Wells et al, 2000).
The separate BRCA1/2 performance measures for Manchester, BRCAPRO and Penn displayed similar trends to the combined values, with the only exception being the use of Penn for assessment of BRCA2 risk, where the positive likelihood ratio was 5.9. A positive result in Penn for BRCA2 mutation should confidently warrant a mutation screen.
Overall, our study suggests that routine use of Penn, Manchester, BRCAPRO or Myriad for predicting BRCA mutation status in clinical practice is not currently justified. Furthermore, as the performance of the models was not influenced by the type of mutation testing, we should consider other sources of error. Risk models are developed and applied on the basis of pedigrees constructed from clinical histories. Yet, these histories are themselves often inaccurate in that cancer verification is often impossible and age at diagnosis is frequently an estimate. We have shown that even where prespecified criteria are established to address the nuances of various patient scenarios, the expert counselor may still interpret the pedigree in a way that alters the risk estimates. BRCA germline mutation testing represents a further source of significant error. The true prevalence of BRCA mutations is often underestimated because of the limitations of molecular testing (sensitivity of molecular techniques 70%) (Eng et al, 2001). Furthermore, models are often derived on the basis of mutation testing results from one uncharacterised individual in a high-risk family. Exclusion of a BRCA mutation in one individual does not necessarily indicate that the family is mutation negative. As uncontrollable factors such as cost, death and unavailability often dictate the choice of individual within a family for mutation testing, it is clear that BRCA mutational status is not a ‘gold standard test’. Given these limitations in developing and applying risk models, we advocate the development of risk prediction models that are less reliant on clinical history.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Antman EM, Grudzien C, Sacks DB (1995) Evaluation of a rapid bedside assay for detection of serum cardiac troponin T. JAMA 273: 1279–1282
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72: 1117–1130
Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, Ponder BJ, Easton DF (2002) A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer 86: 76–83
Barcenas CH, Hosain GM, Arun B, Zong J, Zhou X, Chen J, Cortada JM, Mills GB, Tomlinson GE, Miller AR, Strong LC, Amos CI (2006) Assessing BRCA carrier probabilities in extended families. J Clin Oncol 24: 354–360
Berry DA, Iversen Jr ES, Gudbjartsson DF, Hiller EH, Garber JE, Peshkin BN, Lerman C, Watson P, Lynch HT, Hilsenbeck SG, Rubinstein WS, Hughes KS, Parmigiani G (2002) BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J Clin Oncol 20: 2701–2712
Couch FJ, DeShano ML, Blackwood MA, Calzone K, Stopfer J, Campeau L, Ganguly A, Rebbeck T, Weber BL (1997) BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 336: 1409–1415
de la Hoya M, Diez O, Perez-Segura P, Godino J, Fernandez JM, Sanz J, Alonso C, Baiget M, Diaz-Rubio E, Caldes T (2003) Pre-test prediction models of BRCA1 or BRCA2 mutation in breast/ovarian families attending familial cancer clinics. J Med Genet 40: 503–510
Domchek SM, Eisen A, Calzone K, Stopfer J, Blackwood A, Weber BL (2003) Application of breast cancer risk prediction models in clinical practice. J Clin Oncol 21: 593–601
Eng C, Brody LC, Wagner TM, Devilee P, Vijg J, Szabo C, Tavtigian SV, Nathanson KL, Ostrander E, Frank TS (2001) Interpreting epidemiological research: blinded comparison of methods used to estimate the prevalence of inherited mutations in BRCA1. J Med Genet 38: 824–833
Euhus DM (2001) Understanding mathematical models for breast cancer risk assessment and counseling. Breast J 7: 224–232
Euhus DM, Smith KC, Robinson L, Stucky A, Olopade OI, Cummings S, Garber JE, Chittenden A, Mills GB, Rieger P, Esserman L, Crawford B, Hughes KS, Roche CA, Ganz PA, Seldon J, Fabian CJ, Klemp J, Tomlinson G (2002) Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 94: 844–851
Evans DG, Eccles DM, Rahman N, Young K, Bulman M, Amir E, Shenton A, Howell A, Lalloo F (2004) A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J Med Genet 41: 474–480
Evans DG, Lalloo F, Wallace A, Rahman N (2005) Update on the Manchester Scoring System for BRCA1 and BRCA2 testing. J Med Genet 42: e39
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE (1994) Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 343: 692–695
Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC (2002) Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10 000 individuals. J Clin Oncol 20: 1480–1490
Garvin AM (1998) A complete protein truncation test for BRCA1 and BRCA2. Eur J Hum Genet 6: 226–234
Hartmann LC, Schaid DJ, Woods JE, Crotty TP, Myers JL, Arnold PG, Petty PM, Sellers TA, Johnson JL, McDonnell SK, Frost MH, Jenkins RB (1999) Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 340: 77–84
Hartmann LC, Sellers TA, Schaid DJ, Frank TS, Soderberg CL, Sitta DL, Frost MH, Grant CS, Donohue JH, Woods JE, McDonnell SK, Vockley CW, Deffenbaugh A, Couch FJ, Jenkins RB (2001) Efficacy of bilateral prophylactic mastectomy in BRCA1 and BRCA2 gene mutation carriers. J Natl Cancer Inst 93: 1633–1637
Huang P, Leary J, Smith M, Kirk J (2002) Determining the probability of finding a breast cancer susceptibility gene mutation in clinic based families. In Fifth Australasian Conference on Familial and Genetic Aspects of Cancer, Anon (ed). South Australia: Barossa Valley
James PA, Doherty R, Harris M, Mukesh BN, Milner A, Young MA, Scott C (2006) Optimal selection of individuals for BRCA mutation testing: a comparison of available methods. J Clin Oncol 24: 707–715
Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, Hudis CA, Ellis NA, Boyd J, Borgen PI, Barakat RR, Norton L, Castiel M, Nafa K, Offit K (2002) Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 346: 1609–1615
Kerlikowske K, Grady D, Rubin SM, Sandrock C, Ernster VL (1995) Efficacy of screening mammography. A meta-analysis. JAMA 273: 149–154
Koch GG, Landis JR, Freeman JL, Freeman Jr DH, Lehnen RC (1977) A general methodology for the analysis of experiments with repeated measurement of categorical data. Biometrics 33: 133–158
Liede A, Karlan BY, Narod SA (2004) Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 22: 735–742
Lynch HT, Watson P, Tinley S, Snyder C, Durham C, Lynch J, Kirnarsky Y, Serova O, Lenoir G, Lerman C, Narod SA (1999) An update on DNA-based BRCA1/BRCA2 genetic counseling in hereditary breast cancer. Cancer Genet Cytogenet 109: 91–98
Marroni F, Aretini P, D'Andrea E, Caligo MA, Cortesi L, Viel A, Ricevuto E, Montagna M, Cipollini G, Ferrari S, Santarosa M, Bisegna R, Bailey-Wilson JE, Bevilacqua G, Parmigiani G, Presciuttini S (2004) Evaluation of widely used models for predicting BRCA1 and BRCA2 mutations. J Med Genet 41: 278–285
Meijers-Heijboer H, van Geel B, van Putten WL, Henzen-Logmans SC, Seynaeve C, Menke-Pluymers MB, Bartels CC, Verhoog LC, van den Ouweland AM, Niermeijer MF, Brekelmans CT, Klijn JG (2001) Breast cancer after prophylactic bilateral mastectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 345: 159–164
Myriad (2004) Mutation prevalence tables for BRCA1/2 genes. http://www.myriadtests.com/provider/mutprev.htm (accessed 20 November 2005)
NBCC Genetics Working Group (2000) Advice about familial aspects of breast cancer and ovarian cancer – a guide for health professionals. http://www.genetics.com.au/pdf/pubs/boguidelines.pdf (accessed 20 November 2005)
Nelson HD, Huffman LH, Fu R, Harris EL (2005) Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 143: 362–379
Parmigiani G, Berry D, Aguilar O (1998) Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet 62: 145–158
Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 91: 943–949
Puget N, Sinilnikova OM, Stoppa-Lyonnet D, Audoynaud C, Pages S, Lynch HT, Goldgar D, Lenoir GM, Mazoyer S (1999) An Alu-mediated 6-kb duplication in the BRCA1 gene: a new founder mutation? Am J Hum Genet 64: 300–302
Rebbeck TR, Friebel T, Lynch HT, Neuhausen SL, van't Veer L, Garber JE, Evans GR, Narod SA, Isaacs C, Matloff E, Daly MB, Olopade OI, Weber BL (2004) Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 22: 1055–1062
Rebbeck TR, Levin AM, Eisen A, Snyder C, Watson P, Cannon-Albright L, Isaacs C, Olopade O, Garber JE, Godwin AK, Daly MB, Narod SA, Neuhausen SL, Lynch HT, Weber BL (1999) Breast cancer risk after bilateral prophylactic oophorectomy in BRCA1 mutation carriers. J Natl Cancer Inst 91: 1475–1479
Struewing JP, Watson P, Easton DF, Ponder BA, Lynch HT, Tucker MA (1995) Prophylactic oophorectomy in inherited breast/ovarian cancer families. J Natl Cancer Inst Monogr 17: 33–35
Tulinius H, Olafsdottir GH, Sigvaldason H, Arason A, Barkardottir RB, Egilsson V, Ogmundsdottir HM, Tryggvadottir L, Gudlaugsdottir S, Eyfjord JE (2002) The effect of a single BRCA2 mutation on cancer in Iceland. J Med Genet 39: 457–462
University of Pennsylvania Abramson Cancer Center (2005) The Penn II BRCA1 and BRCA2 Mutation Risk Evaluation Model. http://acgh.afcri.upenn.edu/cgi-bin/bcm.pl (accessed 20 November 2005)
Wells PS, Anderson DR, Ginsberg J (2000) Assessment of deep vein thrombosis or pulmonary embolism by the combined use of clinical model and noninvasive diagnostic tests. Semin Thromb Hemost 26: 643–656
Woodward AM, Davis TA, Silva AG, Kirk JA, Leary JA (2005) Large genomic rearrangements of both BRCA2 and BRCA1 are a feature of the inherited breast/ovarian cancer phenotype in selected families. J Med Genet 42: e31
Acknowledgements
We thank Heather Thorne and Eveline Niedermayer for supply of data from kConFab for this project. We are grateful to the physicians, surgeons and oncologists who endorsed this project, the interviewing staff and the many families who participated in this research. kConFaB has been funded by the Kathleen Cuningham Foundation, National Breast Cancer Foundation, National Health and Medical Research Council (NHMRC), Cancer Council of Victoria, Cancer Council of South Australia, Queensland Cancer Fund, Cancer Council of New South Wales, Cancer Foundation of Western Australian and Cancer Council of Tasmania. This work is supported by National Health and Medical Research Council.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
This work is original. It has been presented by Rachel Williams at the following conference: Familial Cancer 2005: Research and Practice, Couran Cove Qld, August 2005.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kang, H., Williams, R., Leary, J. et al. Evaluation of models to predict BRCA germline mutations. Br J Cancer 95, 914–920 (2006). https://doi.org/10.1038/sj.bjc.6603358
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603358
Keywords
This article is cited by
-
Breast MRI texture analysis for prediction of BRCA-associated genetic risk
BMC Medical Imaging (2020)
-
Validation of a digital identification tool for individuals at risk for hereditary cancer syndromes
Hereditary Cancer in Clinical Practice (2019)
-
Comparison between CaGene 5.1 and 6.0 for BRCA1/2 mutation prediction: a retrospective study of 150 BRCA1/2 genetic tests in 517 families with breast/ovarian cancer
Journal of Human Genetics (2017)
-
Breast cancer risk models: a comprehensive overview of existing models, validation, and clinical applications
Breast Cancer Research and Treatment (2017)
-
Evaluation of the Dutch BRCA1/2 clinical genetic center referral criteria in an unselected early breast cancer population
European Journal of Human Genetics (2015)
