
Abstract
The serine/threonine kinase, mTOR (mammalian Target of Rapamycin) has become a focus for cancer drug development. Rapamycins are highly specific inhibitors of mTOR and potently suppress tumour cell growth by retarding cells in G1 phase or potentially inducing apoptosis. Currently, both rapamycin and several analogues are being evaluated as anticancer agents in clinical trials. Results indicate that many human cancers have intrinsic resistance and tumours initially sensitive to rapamycins become refractory, demonstrating acquired resistance. Here, we consider mechanisms of resistance to inhibitors of mTOR.
Similar content being viewed by others


Main
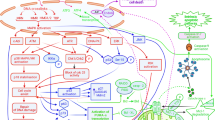
The mammalian target of rapamycin, mTOR is a serine/threonine kinase evolutionarily conserved from yeast to human in the catalytic domain. As the C-terminus of mTOR is highly homologous to the catalytic domain of phosphatidylinositol 3 kinase (PI3K), mTOR is considered a member of PI3K-related kinase family (designated PIKK). The role of mTOR in cell growth, metabolism, and cancer has been reviewed extensively (Bjornsti and Houghton, 2004; Wullschleger et al, 2006). Signalling pathways upstream and downstream of mTOR are presented in Figure 1. Increasing evidence has implicated mTOR as a sensor that integrates extracellular and intracellular events, coordinating cell size (growth), proliferation, and survival. Mammalian target of rapamycin may directly or indirectly regulate translation initiation (through the mTOR-raptor [TORC1] complex), actin organisation (through the mTOR-rictor [TORC2] complex), membrane trafficking, protein degradation, ribosome biogenesis and tRNA synthesis, as well as transcription. Mammalian target of rapamycin signalling is negatively regulated by amino-acid deficiency and increased AMP levels, suppressing cap-dependent protein synthesis. Thus, mTOR in concert with tuberin and hamartin proteins that form the tuberous sclerosis complex (TSC) and Rheb, appears to sense nutrient and growth factor status, as well as energy charge, and regulates progression from G1 to S phase.

Chemical structure of rapamycin and its analogues currently in clinical trials as anticancer chemotherapeutic agents. Bars indicate the chemical modifications to rapamycin.
Clinical development of rapamycin analogues
A number of clinical trials with the rapalogues (CCI-779, RAD001, and AP23573; Figure 1) have now been completed. The phase I trial evaluating the safety of CCI-779 was examined for both daily and weekly i.v. treatment in patients with a number of different types of tumours (Dancey, 2002). Objective responses (non-small-cell carcinoma, neuroendocrine, and breast carcinomas), and minor responses or stable disease (cervical carcinoma, uterine carcinoma, renal cell carcinoma, and soft tissue sarcoma) were observed. Phase II trials for renal cell carcinoma yielded an objective response rate of 5–7%, a minor response rate of 26–29%, and stable disease in approximately 40% of the patients (Atkins et al, 2004). Treatment is associated with an increase in survival time of approximately 4 months. For Mantle cell lymphoma, an overall response rate of 38% (3% complete response and 35% partial response), with a median time to progression of 6.9 months in responders vs 6.5 months for all patients treated (Witzig et al, 2005). CCI-779 appears to have significant activity in endometrial cancer, irrespective of PTEN (phosphatase and tensin homologue deleted on chromosome ten) status, yielding objective responses in 25% of patients, and causing disease stabilisation in over half of the patients on study. Median progression-free survival was 8 months. For AP23573, phase II results suggest significant activity in approximately 30% of patients with previously treated subtypes of sarcoma. Thus, in these studies there is clear evidence of acquired resistance where tumour growth is initially arrested but where relapse occurs on treatment with rapamycin analogues. In contrast, the majority of patients with advanced breast cancer (overall response rate of 9.2%) appear to exhibit intrinsic resistance to this class of agent (Chang et al, 2005).
Mechanism of action of rapamycins
Rapamycin and its analogues in clinical development are highly specific inhibitors of mTOR, and differ only slightly in chemical structure (Figure 1) that results in improved chemical stability and pharmaceutical properties. Rapamycins bind a Mr 12 000 cytosolic immunophilin termed FK-binding protein (FKBP-12). The FKBP–rapamycin complex binds to the FK–rapamycin binding (FRB) domain of mTOR, resulting in inhibition of the function of mTOR in the TORC1 complex. Whether rapamycin or FKBP–rapamycin complex directly inhibits the kinase activity of mTOR is controversial, rather FKBP–rapamycin complex may disrupt higher order mTOR–protein complexes, perhaps displacing substrates from the catalytic domain (Bjornsti and Houghton, 2004). As each of the rapalogues in clinical development has an essentially identical mechanism of action, it is probable that resistance mechanisms will be similar.
Rapamycin-sensitive signalling pathways mediated by mTOR
4E-BP1 and S6K1 are the best characterised downstream effector molecules of mTOR. 4E-BP1 functions as a suppressor of eukaryotic initiation factor 4E (eIF4E), the mRNA cap-binding protein. In response to mitogens, six sites of 4E-BP1 can be phosphorylated (Shah et al, 2000). In the presence of rapamycin, 4E-BP1 becomes hypo-phosphorylated and associates with eIF4E, preventing binding of eIF4E to the scaffold protein eIF4G and formation of the eIF4F pre-initiation complex required for cap-dependent translation of mRNA. As a result, rapamycin may downregulate mTOR-controlled synthesis of essential proteins involved in cell cycle progression, such as cyclin D1 and ornithinine decarboxylase, and survival (hypoxia inducible factor 1α, and c-MYC).
S6K1 and S6K2, downstream of mTOR, are both inhibited by rapamycin (Jefferies et al, 1997). Similar to 4E-BP1, activation of S6K1 requires hierarchical phosphorylation. In vitro, mTOR phosphorylates only Thr389 in the regulatory domain. A recent model proposed by the Blenis laboratory suggests that S6K1 is phosphorylated in the pre-initiation complex by mTOR leading to dissociation and full activation of S6K1 by PDK1. Activated S6K1 phosphorylates eIF4B, which then binds in the pre-initiation complex (Holz et al, 2005).
Regulation of sensitivity to mTOR inhibition by signalling pathways
Phosphatase and tensin homologue deleted on chromosome ten (PTEN) is a dual-specificity protein phosphatase, and functions as a major negative regulator of the PI3-kinase/Akt signalling pathway. Loss of PTEN occurs frequently in human tumours (Vivanco and Sawyers, 2002), resulting in hyperphosphorylation or activation of Akt. Activated Akt positively regulates mTOR-dependent pathways through inhibition of the TSC complex, and forms the basis for rapamycin hypersensitivity of PTEN-deficient or -mutant cells. Either loss of PTEN or activation of Akt resulted in an increased sensitivity to the rapalogue CCI-779 in a panel of brain, prostate, breast cancer cell lines, and human tumours implanted in athymic nude mice (Yu et al, 2001). Most CCI-779-resistant tumour cells had a low or moderate level of activated Akt, although there are exceptions where loss of PTEN is not associated with hypersensitivity to CCI-779. In contrast to CCI-779 resistant lines, sensitive breast cancer cell lines were estrogen-dependent or lacked expression of the tumour suppressor PTEN, and/or overexpressed the Her-2 oncogene. One proposed mechanism by which low Akt activity induces rapamycin resistance is through allowing continued cap-independent protein synthesis of cyclin D1 and c-MYC proteins. The cyclin D1 mRNA 5′ untranslated region contains an internal ribosome entry site (IRES) and both this IRES and the c-MYC IRES are negatively regulated by Akt activity, and the function of both IRES's is enhanced following exposure to rapamycin. Hence, continued IRES-mediated translation initiation may permit cell cycle progression upon mTOR inactivation in cells in which Akt kinase activity is relatively low (Shi et al, 2005). Thus, one biomarker for intrinsic rapamycin resistance may be low levels of activation of Akt. On the other hand, rapamycin sensitivity of breast cancer lines correlated inversely with phospholipase D (PLD) activity. Elevating PLD activity led to rapamycin resistance, and inhibition of PLD activity increased rapamycin sensitivity. This is consistent with the notion that PLD-generated phosphatidic acid competes for rapamycin–FKBP binding to mTOR. In yeast, activation of the RAS/cAMP signalling pathway also confers pronounced resistance to rapamycin (Schmelzle et al, 2004); however, this association has not been reported for mammalian cells.
Mutations in FKBP12 and mTOR
Although direct inhibitors of mTOR kinase activity are being developed, most data on mechanisms of resistance apply to rapamycins. Rapamycin first binds FKBP-12 in mammalian cells. Either specific mutations in FKBP12 that prevent the formation of FKBP–rapamycin complex, or mutations in the FRB domain of mTOR that decrease the affinity of binding of the FKBP–rapamycin complex would cause rapamycin resistance. Such mutations were first found in the yeast Saccharomyces cerevisiae, where deletion of the RBP1 gene (a homologue of mammalian FKBP-12) resulted in recessive resistance to rapamycin, and expression of RBP1 restored rapamycin sensitivity. In mouse mast cells, mutations in FKBP-12 that substantially reduced binding affinity of FKBP-12 for rapamycin, rendered rapamycin resistance (Fruman et al, 1995). Resistance to rapamycin selected after mutagenesis is related to a dominant phenotype consistent with mutation in the FRB domain of mTOR (Dumont et al, 1995). Expression of an FRB-mutant mTOR (Ser2035 → Ile), having reduced affinity for binding the FKBP–rapamycin complex, confers high-level resistance (Dumont et al, 1995), Figure 2A.

(A) Predicted mechanisms of resistance to rapamycin analogues. Rapamaycin or its derivatives (red balls) can be eliminated from cells by ABC transporters such as P-glycoprotein. Mutations of FKBP or mTOR (yellow stars) confer resistance. Acquired resistance to rapamycin has been associated with decreased stoichiometry between 4E-BP and eIF4E, either through decreased translation of 4E-BP or overexpression of eIF4E. (B) Inhibition of mTOR leads to decreased translation of cyclin D1 mRNA, and reduced levels of cyclin D1. In many cells, there is a concomitant stabilisation of the cyclin-dependent kinase inhibitor p27Kip1, and inhibition of CDK-cyclin activity, and decreased phosphorylation of RB. Cells deficient in p27Kip1 are partially resistant, whereas RB-null cells are completely resistant to inhibition of proliferation by rapamycin.
Dysregulation of EIF4E
4E-BP1, the suppressor of eIF4E, has been widely recognised as the other primary downstream effector of mTOR. Acquired resistance to rapamycin was associated with decreased levels (∼10-fold) of 4E-BP1, the suppressor of eIF4E (Dilling et al, 2002) owing to decreased translation of 4E-BP1. In cells that reverted to rapamycin sensitivity, total levels of 4E-BP1 became similar to those in parental cells, and 4E-BP1 bound to eIF4E had similar response to serum starvation and IGF-I stimulation as found in parental cells. These data suggest that decrease of 4E-BP1 expression results in dysregulation of eIF4E, conferring rapamycin resistance.
These results indicate that the rapamycin-regulated eIF4E pathway is crucial in inducing growth arrest, and dysregulation of eIF4E may facilitate the malignant phenotype. This is supported by clinical observations that dysregulation of the eIF4E pathway is associated with tumour progression (Nathan et al, 1997). In addition to decreased 4E-BP1 expression, increased eIF4E expression in mice is tumorigenic and increases the rate of tumour progression. When 4E-BP1 is overexpressed in highly resistant HCT8 cells (IC50>10 000 ng ml−1), they become sensitive (IC50 <10 ng ml−1) to rapamycin (Dilling et al, 2002). Potentially, the ratio of 4E-BP to eIF4E may determine whether inhibition of mTOR elicits a biologically significant tumour response (Figure 2A).
Defective regulation of the retinoblastoma (RB) protein checkpoint
In malignant cells, the RB checkpoint can be bypassed by loss of RB protein or function, loss of p27Kip1, and overexpression of cyclin E or other cyclins. The role of p27Kip1 in modulating rapamycin sensitivity (Luo et al, 1996) implies that mTOR has a role in controlling the phosphorylation state of RB, however, this may be cell-context specific. Rapamycin prevents mitogen-stimulated downregulation of the cdk inhibitor p27Kip1 and suggests that p27Kip1 is involved in the antiproliferative activity of rapamycin. Prolonged culture of sensitive cells in the presence of drug resulted in rapamycin-resistant clones that exhibited low p27Kip1 protein, no decrease in mRNA level but a high rate of ubiquitin-independent degradation. p27Kip1 in resistant cells could not be regulated either in response to serum or rapamycin, and as a result, RB phosphorylation was not blocked. However, only partial resistance to antiproliferative activity of rapamycin was found in p27−/− mouse embryo fibroblasts (MEFs) and p27−/− splenic T lymphocytes, indicating that there exist p27-dependent and -independent pathways that determine rapamycin sensitivity (Figure 1B). Rapamycin also inhibits insulin-induced RB phosphorylation in 3T3-L1 adipocytes but not in rapamycin-sensitive NIH3T3 fibroblasts where rapamycin does not alter the phosphorylation status of RB or two RB-related proteins p107 and p130. Similarly, in Ba/F3 p210 cells expressing BCR/ABL, rapamycin arrested growth, but failed to alter the hyperphosphorylation of RB protein (Gesbert et al, 2000). RB-null MEFs, however, are highly resistant to rapamycin (IC50 >10 μg ml−1) relative to wild-type cells (IC50∼1 ng ml−1) (unpublished data) (Figure 2B).
Defective regulation of the p53 checkpoint
p53 is a tumour suppressor and transcription factor that has been found to be mutated in ∼50% of human cancers and p53 status may also determine the cellular response to rapamycin (Huang et al, 2003). When cultured under autocrine conditions (serum-free) p53+/+ MEFs exposed to rapamycin accumulate in G1 phase and maintain viability, whereas p53-mutant cells accumulate in G1 phase and undergo apoptosis. In p53-defective cells, inhibition of mTOR leads to a rapid and maintained induction of a stress response characterised by increased phospho-c-Jun, whereas in cells with functional p53 this stress response is suppressed (Huang et al, 2003). Mechanistically, inhibition of mTOR leads to decreased activity of protein phosphatase 5 (PP5) and activation of apoptosis signal regulating kinase 1. Rapamycin-induced apoptosis can be inhibited by expression of functional p53, by overexpression of PP5, decreased expression of 4E-BP1, or overexpression of Bcl2.
Mutations of PP2A-related protein phosphatases
Several serine/threonine protein phosphatases have been identified as the components of mTOR signalling pathway in mammalian cells (Dennis et al, 1999). Rapamycin resistance caused by mutations of PP2A-related phosphatases was first found in yeast. Studies in mammalian cells also indicate that association of α4 (the homologue of yeastTap42) with PP2A, PP4, and PP6 is related to rapamycin sensitivity (Murata et al, 1997). For example, in rapamycin-sensitive Jurkat cells, rapamycin dissociated α4 from PP2Ac, and transfection of mouse α4 into Jurkat cells conferred rapamycin resistance. In rapamycin-resistant Raji cells, rapamycin did not affect association of α4 with PP2Ac. However, other studies have not demonstrated rapamycin-induced dissociation of α4 from PP2A or PP6. Thus, at this time the significance of α4 to rapamycin resistance is controversial.
Mutations of ATM
ATM (ataxia telangiectasia, mutated) cells have been reported to be rapamycin resistant. In contrast, early passage murine embryo fibroblasts derived from ATM−/− mice are not resistant to rapamycin (Germain and Houghton, unpublished data) distinguishing the null phenotype from the ATM mutant cells. As ATM cells have genetic instability, it is possible that resistance to rapamycin is a consequence of additional mutations and not related directly to defects in ATM signalling.
Mutations of 14-3-3
In yeast, the two homologues of the mammalian 14-3-3 proteins act as multicopy suppressors of the growth-inhibitory phenotype caused by rapamycin (Bertram et al, 1998). Overexpression of either BMH1 or BMH2, or three human 14-3-3 isoforms (β, τ, and η) in yeast conferred rapamycin resistance, whereas disruption sensitised to rapamycin. The mechanism by which 14-3-3 proteins cause rapamycin resistance is not known; however, potentially 14-3-3 proteins may play a role in determining rapamycin sensitivity in mammalian cells.
Prevention of apoptosis by growth factors and cytokines
Substantial data support a role for mTOR signalling through the eIF4E pathway in promoting cell survival, although exogenous factors may modulate the apoptotic response to mTOR inhibition. IGF-I or insulin protect from rapamycin-induced apoptosis (Kurmasheva and Houghton, 2006) by activating Akt-dependent and -independent pathways resulting in phosphorylation and inactivation of the proapoptotic protein Bad, thus promoting the activity of Bcl2. Overexpression of Bcl2 also confers resistance to rapamycin-induced cell killing.
Anecdotally, in many cell lines and patient tumour biopsies (O’Reilly et al, 2006), inhibition of mTOR activates Akt and inhibits downstream substrates (FoxO transcription factors, Bad, and GSK3α/β). Increased phospho-Akt was correlated with increased levels of IRS-1 but not IRS-2. IRS-1 is phosphorylated by S6K1 at Ser312 resulting in its dissociation from the IGF-1 receptor, and proteasome-mediated degradation (Shi et al, 2005). Inhibition of mTOR inactivates S6K1 and stabilises IRS-1 associated with receptors, thus alleviating this negative regulation. Inhibition of IGF-1R abrogated rapamycin-induced phospho-Akt induction and substrate phosphorylation, and combination of rapamycin with an inhibitor of IGF-1R had additive effects on proliferation irrespective of PTEN status and increased apoptosis in a PTEN mutant cancer cell line (O’Reilly et al, 2006). Thus, either exogenous IGF-1 or derepression of IRS-1 in the presence of rapamycin may induce resistance to growth inhibition or apoptosis induced by mTOR inhibition (see Easton et al, 2006). Similarly IL-3 induces expression of Pim-2 and confers resistance to rapamycin, probably through the dual action of phosphorylating 4E-BP1 and Bad. Pim kinases are components of the transcriptional response to cytokines or antigen receptor ligation, and overexpression of Pim-1 or Pim-2 can maintain 4E-BP1 phosphorylation (Ser65) in the presence of rapamycin, conferring resistance. Consistent with the role of Pim kinases modulating the activity of mTOR inhibitors, Pim-2−/− or Pim-1−/−Pim-2−/− cells are hypersensitive to rapamycin (Fox et al, 2005).
Inhibition of mTOR in TORC2 complexes
Mammalian target of rapamycin complexed with rictor (TORC2) is thought to be insensitive to rapamycin, and to play a role in regulating the actin cytoskeleton (Wullschleger et al, 2006). The TORC2 complex phosphorylates Akt at Ser473, and consequently may regulate both survival through Akt, and also TORC1 (mTOR-raptor) activity. Prolonged inactivation of TORC1 by rapamycin potentially leads to a shift of mTOR from the TORC2 complex to being rapamycin-bound, and lead to decreased Akt activation, activation of GSK-3β (which regulates cyclin D1 proteolysis, and enhances rapamycin-mediated growth inhibition), and apoptosis. Thus, for direct inhibitors of mTOR kinase, the potential for upregulation of Akt via the IRS-1 pathway and protection from cell death seems less likely. The TORC2 complex will also be targeted by direct mTOR kinase inhibitors that bind into the ATP pocket of the catalytic domain. By analogy to acquired resistance mechanisms to imatinib or gefitinib that frequently are caused by mutations in ATP-binding site for BCR-ABL and ERBB1, respectively, it is probable that ATP-mimetic mTOR inhibitors will select for binding site mutants.
ABC-Transporters
The ability of cyclosporin A, FK506, and rapamycin to overcome P-glycoprotein-mediated multidrug resistance has been demonstrated (Arceci et al, 1992). The multidrug resistance-reversing activity increased in the order rapamycin <FK506 < cyclosporin A, irrespective of whether the resistant cells overexpressed hamster or human P-glycoprotein. The interaction of the three macrolides with P-glycoprotein was characterised by their ability bind with high affinity to P-glycoprotein, and to competitively inhibit the photoaffinity labelling of plasma membranes of drug-resistant cells by iodomycin. Although an inhibitor of P-glycoprotein activity rapamycin decreased the rate of ATP hydrolysis with respect to the basal rate, it did not completely inhibit the activity. Hence, rapamycin can be classified as a substrate for transport by P-glycoprotein (Kerr et al, 2001), as a consequence tumours overexpressing P-glycoprotein (MDR1) may be resistant to rapamycins (Figure 1A).
Angiogenesis-associated resistance mechanisms
Guba et al (2002) first reported the anecdotal result where cells were resistant to rapamycin in vitro, but tumours from the same cells in mice were growth inhibited by rapamycin treatment. They concluded that the antitumor activity of rapamycin was due to its antiangiogenic activity as rapamycin decreased production of vascular endothelial growth factor (VEGF) and markedly inhibited response of vascular endothelial cells to stimulation by VEGF. Similar results have been obtained with human cancer cell lines that are intrinsically resistant to rapamycin analogues in vitro, but where in vivo growth of tumour xenografts is inhibited significantly. The mTOR pathway controls cap-dependent translation, and hence potentially levels of hypoxia inducible factor 1α (HIF-1α), cellular proliferation, and migration. Thus, inhibition of mTOR may have direct effects on cancer cell proliferation and survival, indirect effects via inhibition of HIF-1α, thus reducing tumour-elicited VEGF, direct effects on vascular endothelial cells, or vascular smooth muscle cells (Humar et al, 2002; Majumder et al, 2004). For example, induction of HIF-1α and VEGF by the CML-associated oncogene, BCR-ABL, is mTOR-dependent (Mayerhofer et al, 2002), and in vitro, rapamycin inhibited VEGF production in primary cultures from BCR-ABL transformed, imatinib resistant, CML (Mayerhofer et al, 2005). The role for mTOR in VEGF production is supported by regulation of HIF-1α by mTOR signalling and increased VEGF in cells deficient in the TSC that negatively regulates mTOR via Rheb (Hudson et al, 2002). However, other studies support a role mainly for PI3K and to a lesser extent mTOR being required for insulin-induced HIF-1α expression (Treins et al, 2002). Our studies indicate that rapamycin treatment has little effect on hypoxia-driven VEGF production in most rhabdomyosarcoma or neuroblastoma cell lines (Kurmasheva et al, submitted). Thus, in these cells it is unlikely that rapamycin would block tumour-derived VEGF, although it may directly block the response of vascular endothelial or other stromal cells in tumour tissue. Potentially, in vivo resistance to mTOR inhibition could be elicited by secretion of angiogenic factors that signal to stromal cells via mTOR-independent pathways to increase proliferation or motility of vascular cells.
Conclusions
The mechanisms for resistance to rapamycin analogues are likely to be complex. In part, it remains unclear exactly why inhibition of mTOR signalling results in growth inhibition, as inhibition of mTOR signalling appears similar in rapamycin-sensitive or -resistant cell lines. It is also probable that resistance mechanisms will be similar for the rapalogues currently in clinical development. The mechanism(s) for resistance to catalytic kinase inhibitors, however may more likely relate to mTOR mutations within the ATP binding site as identified for imatinib and gefitinib. Although preclinical models can suggest potential mechanisms for resistance to rapamycins, it is of critical importance to characterise tumour biopsy tissue at initiation of treatment and at subsequent relapse in order to ascertain which mechanisms apply to clinical cancer.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arceci RJ, Stieglitz K, Bierer BE (1992) Immunosuppressants FK506 and rapamycin function as reversal agents of the multidrug resistance phenotype. Blood 80: 1528–1536
Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G, Sherman ML (2004) Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 22: 909–918
Bertram PG, Zeng C, Thorson J, Shaw AS, Zheng XF (1998) The 14-3-3 proteins positively regulate rapamycin-sensitive signaling. Curr Biol 8: 1259–1267
Bjornsti MA, Houghton PJ (2004) The TOR pathway: a target for cancer therapy. Nat Rev Cancer 4: 335–348
Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Tham YL, Kalidas M, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, Lewis MT, Wong H, O’Connell P (2005) Patterns of resistance and incomplete response to docetaxel by gene expression profiling in breast cancer patients. J Clin Oncol 23: 1169–1177
Dancey JE (2002) Clinical development of mammalian target of rapamycin inhibitors. Hematol Oncol Clin North Am 16: 1101–1114
Dennis PB, Fumagalli S, Thomas G (1999) Target of rapamycin (TOR): balancing the opposing forces of protein synthesis and degradation. Curr Opin Genet Dev 9: 49–54
Dilling MB, Germain GS, Dudkin L, Jayaraman AL, Zhang X, Harwood FC, Houghton PJ (2002) 4E-binding proteins, the suppressors of eukaryotic initiation factor 4E, are down-regulated in cells with acquired or intrinsic resistance to rapamycin. J Biol Chem 277: 13907–13917
Dumont FJ, Staruch MJ, Grammer T, Blenis J, Kastner CA, Rupprecht KM (1995) Dominant mutations confer resistance to the immunosuppressant, rapamycin, in variants of a T cell lymphoma. Cell Immunol 163: 70–79
Easton JB, Kurmasheva RT, Houghton PJ (2006) IRS-1: auditing the effectiveness of mTOR inhibitors. Cancer Cell 9: 153–155
Fox CJ, Hammerman PS, Thompson CB (2005) The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med 201: 259–266
Fruman DA, Wood MA, Gjertson CK, Katz HR, Burakoff SJ, Bierer BE (1995) FK506 binding protein 12 mediates sensitivity to both FK506 and rapamycin in murine mast cells. Eur J Immunol 25: 563–571
Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD (2000) BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem 275: 39223–39230
Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8: 128–135
Holz MK, Ballif BA, Gygi SP, Blenis J (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580
Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ (2003) Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1). Mol Cell 11: 1491–1501
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22: 7004–7014
Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ (2002) Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J 16: 771–780
Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G (1997) Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J 16: 3693–3704
Kerr KM, Sauna ZE, Ambudkar SV (2001) Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J Biol Chem 276: 8657–8664
Kurmasheva RT, Houghton PJ (2006) IGF-I mediated survival pathways in normal and malignant cells. Biochim Biophys Acta 177: 1–22
Luo Y, Marx SO, Kiyokawa H, Koff A, Massague J, Marks AR (1996) Rapamycin resistance tied to defective regulation of p27Kip1. Mol Cell Biol 16: 6744–6751
Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR (2004) mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 10: 594–601
Mayerhofer M, Aichberger KJ, Florian S, Krauth MT, Hauswirth AW, Derdak S, Sperr WR, Esterbauer H, Wagner O, Marosi C, Pickl WF, Deininger M, Weisberg E, Druker BJ, Griffin JD, Sillaber C, Valent P (2005) Identification of mTOR as a novel bifunctional target in chronic myeloid leukemia: dissection of growth-inhibitory and VEGF-suppressive effects of rapamycin in leukemic cells. FASEB J 19: 960–962
Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C (2002) BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood 100: 3767–3775
Murata K, Wu J, Brautigan DL (1997) B cell receptor-associated protein alpha4 displays rapamycin-sensitive binding directly to the catalytic subunit of protein phosphatase 2A. Proc Natl Acad Sci USA 94: 10624–10629
Nathan CA, Carter P, Liu L, Li BD, Abreo F, Tudor A, Zimmer SG, De Benedetti A (1997) Elevated expression of eIF4E and FGF-2 isoforms during vascularization of breast carcinomas. Oncogene 15: 1087–1094
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508
Schmelzle T, Beck T, Martin DE, Hall MN (2004) Activation of the RAS/cyclic AMP pathway suppresses a TOR deficiency in yeast. Mol Cell Biol 24: 338–351
Shah OJ, Anthony JC, Kimball SR, Jefferson LS (2000) 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab 279: E715–E729
Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J (2005) Cyclin D1 and c-MYC internal ribosome entry site (IRES)-dependent translation is regulated by Akt activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem 280: 10964–10973
Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E (2002) Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem 277: 27975–27981
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-kinase Akt pathway in human cancer. Nat Rev Cancer 2: 489–501
Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, Dakhil SR, Gross H, Kaufmann SH (2005) Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 23: 5347–5356
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471–484
Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, Gibbons JJ (2001) mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer 8: 249–258
Acknowledgements
Original work cited from this laboratory was supported by USPHS grants CA77776, CA96696, CA23099, and CA21675 (Cancer Center Support Grant) from the National Cancer Institute, and by American, Lebanese, Syrian Associated Charities (ALSAC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kurmasheva, R., Huang, S. & Houghton, P. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer 95, 955–960 (2006). https://doi.org/10.1038/sj.bjc.6603353
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603353
Keywords
This article is cited by
-
High-dose rapamycin exerts a temporary impact on T. reesei RUT-C30 through gene trFKBP12
Biotechnology for Biofuels (2021)
-
Direct phosphorylation and stabilization of HIF-1α by PIM1 kinase drives angiogenesis in solid tumors
Oncogene (2021)
-
HPV-16 E7 expression up-regulates phospholipase D activity and promotes rapamycin resistance in a pRB-dependent manner
BMC Cancer (2018)
-
In silico evaluation of DNA Damage Inducible Transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies
Scientific Reports (2017)
-
Targeting mTOR/p70S6K/glycolysis signaling pathway restores glucocorticoid sensitivity to 4E-BP1 null Burkitt Lymphoma
BMC Cancer (2015)
