
Abstract
Mdm2 is frequently overexpressed in sarcoma cells and may contribute to drug resistance by increasing p53 degradation. We investigated the induction of apoptosis in sarcoma cells via adenovirus-mediated gene transfer of wild-type p53 and two modified p53 genes, p53 14/19 and p53 22/23, whose protein products are resistant to Mdm2-mediated degradation. We found that adenovirus-wt p53 (Ad-wt p53) induces significant apoptosis in HT1080 fibrosarcoma cells expressing low levels of Mdm2, but fails to induce apoptosis in SJSA osteosarcoma cells expressing high levels of Mdm2. In contrast, Ad-p53 14/19 induces significant apoptosis in both cell lines. Interestingly, Ad-p53 22/23, a vector encoding a transcription-defective p53 mutant, causes limited apoptosis in both cell lines. We demonstrate that doxorubicin induces phosphorylation of both wt p53 and p53 14/19 protein at multiple sites. We tested the efficacy of doxorubicin and cisplatin with either Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19. SJSA cells, although harbouring endogenous wt p53, did not undergo significant apoptosis following doxorubicin or cisplatin exposure alone or combined with Ad-wt p53. In contrast, doxorubicin or cisplatin plus Ad-p53 14/19 induced significant apoptosis. Gene transfer of p53 14/19 in combination with the administration of doxorubicin or cisplatin is a potential therapeutic approach for cancers expressing high levels of Mdm2.
Similar content being viewed by others


Main
Sarcomas are uncommon malignancies generally arising from connective tissue or bone. Although these neoplasms are rare, important insights have been gained from studying their biology, and have led to new therapeutic approaches (Joensuu et al, 2001). One such discovery is the fundamental role of the p53/Mdm2 pathway.
The p53 tumour suppressor gene encodes a transcription factor that plays a critical role in regulating the cell cycle and maintaining genomic integrity by inducing growth arrest, DNA repair and apoptosis in response to a variety of stresses (Vogelstein and Kinzler, 1992; Donehower and Bradley, 1993; Prives and Hall, 1999). Functional inactivation of p53 is a common event in the development of human cancer (Levine et al, 1991; Vogelstein et al, 2000), and p53 inactivation is typically caused by p53 mutations, Mdm2 overexpression and other mechanisms. It is well known that more than 50% of all human cancers contain p53 mutations. To target the p53 mutations in human cancers, ONYX-015 (dl1520), which selectively replicates in and kills p53-deficient cells, is being explored as an anti-tumour agent in clinical trials (Hann and Balmain, 2003).
Mdm2, a major negative regulator of p53, inactivates p53 protein by binding its transcriptional activation domain, inhibiting p53's regulation of target genes and its antiproliferative effects (Chen et al, 1996; Haupt et al, 1997a; Kubbutat et al, 1997, 1998). Mdm2 is the major p53 E3 ubiquitin ligase that governs ubiquitination and degradation of p53 (Haupt et al, 1997a; Honda et al, 1997; Kubbutat et al, 1997). mdm2 amplification is common in sarcomas, and most of these tumours retain wt p53 (Leach et al, 1993; Keleti et al, 1996), suggesting that tumours with Mdm2 overexpression bypass the need to mutate p53.
Loss of p53 function contributes not only to tumour progression but also to resistance of tumours to chemotherapy or radiation therapy (Lowe et al, 1993). Overexpression of Mdm2 can confer resistance to cytotoxic drugs (Kondo et al, 1995, 1996; Suzuki et al, 1998). Mdm2 has also been shown to induce expression of the multidrug resistance 1 (mdr1) gene and its main product P-glycoprotein (P-gp) (Kondo et al, 1996), which has been implicated in chemoresistance in human sarcomas (Chan et al, 1990; Hoffman et al, 1999; Jiminez et al, 1999). Wild-type p53 can sensitise sarcoma cells harbouring p53 mutations to doxorubicin by downregulating MDR-1 and P-gp expression (Zhan et al, 2001).
Restoration of normal p53 function has been evaluated as a strategy for cancer therapy (Beaudry et al, 1996). The most direct approach involves transfer of wt p53 gene to cancer cells that lack endogenous p53 function (Yang et al, 1995; Zhang et al, 1995; Ko et al, 1996). However, since many sarcomas overexpress Mdm2, and high levels of Mdm2 inactivate and cause degradation of p53 (Haupt et al, 1997a; Momand et al, 1992; Oliner et al, 1992, 1993; Nakayama et al, 1995; Momand et al, 1998; Vogelstein et al, 2000), wt p53 transfer into sarcoma cells that overexpress Mdm2 is unlikely to be efficacious (Chen et al, 1996; Meng et al, 1998).
To overcome the inhibition of p53 by Mdm2, two modified p53 genes were constructed, p53 14/19 and p53 22/23, which contain double substitutions at residues Leu-14; Phe-19, and Leu-22; Trp-23 (Lin et al, 1994, 2000). As p53 14/19 and p53 22/23 proteins are defective in the ability to bind Mdm2, they are completely resistant to Mdm2-mediated degradation (Chen et al, 1995, 1996; Haupt et al, 1997a). In this study, we evaluate induction of apoptosis by adenovirus-p53 14/19 (Ad-p53 14/19) and adenovirus-p53 22/23 (Ad-p53 22/23) in low and high Mdm2 sarcoma cell lines. The previous study indicated that high levels of Mdm2 might confer multidrug resistance (Cocker et al, 2001). Therefore, we also investigated the synergistic effect on apoptosis between Ad-wt p53 or Ad-p53 14/19 and doxorubicin or cisplatin. We further examined the difference in transactivation function between Ad-p53 14/19 and Ad-wt p53 when combined with doxorubicin in an Mdm2 overexpressing sarcoma cell line.
Materials and methods
Cell lines and cell culture
SJSA osteosarcoma cells (provided by Jiandong Chen, H Lee Moffitt Cancer Center), HT1080 fibrosarcoma cells (American Type Culture Collection, Manassas, VA, USA) and normal human skin fibroblasts (NHF) that have a limited life span (provided by Mats Ljungman, University of Michigan Cancer Center) were used. SJSA cells exhibit amplification of mdm2 and express high levels of Mdm2 protein (Oliner et al, 1992; Landers et al, 1997), whereas HT1080 cells express low levels of Mdm2 (Lin et al, 2000). Both cell lines express low levels of wt p53 (Evdokiou and Cowled, 1998). Normal human skin fibroblasts were used as a control to assess the safety of adenovirus-mediated transfer of p53 14/19 in vitro. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco/BRL, Grand Island, NY, USA) containing 10% foetal bovine serum (FBS) and antibiotics (penicillin G 5000 U ml−1, streptomycin 5000 μg ml−1, Gibco/BRL). Cells were grown as an attached monolayer at 37°C in a humidified atmosphere with 5% CO2.
Adenovirus p53 vector, doxorubicin and cisplatin
To generate the recombinant adenovirus-p53 vectors, as described previously (Lin et al, 2000), cDNA for wt p53, p53 14/19 or p53 22/23 was cloned into an adenovirus vector, pACCMVpLpA(-)loxD (University of Michigan Vector Core). The negative control adenovirus (referred to as NCV, pACCMVpLpA(-)loxD) contains the same backbone as the other constructs. The human cytomegalovirus promoter was used to drive p53 transcription for high level, constitutive expression. Doxorubicin (Calbiochem®, CN Biosciences, San Diego, CA, USA) was dissolved in water to produce a stock solution of 0.5 mg ml−1 and stored at 4°C. Cisplatin (American Pharmaceutical Partners, INS. Los Angeles) was diluted 1 mg per ml with 9 mg sodium chloride per ml in sterile water, and stored at room temperature.
Apoptosis assay
To quantify apoptosis, 5 × 105 cells/10-cm dish were infected by mock (no) infection, NCV, Ad-wt p53, Ad-p53 14/19 or Ad-p53 22/23 at a multiplicity of infection of 100–200 plaque forming units/cell in DMEM containing 2% FBS. The next day, the medium was removed and cells were rinsed twice with phosphate-buffered saline (PBS) to remove the adenovirus. Cells were then incubated in DMEM containing 10% FBS for 48 h. To assess synergy between Ad-wt p53 or Ad-p53 14/19 and doxorubicin or cisplatin, some cells were also treated with 0.1 μg ml−1 doxorubicin or 5 μg ml−1 cisplatin, respectively. At 72 h after infection, both adherent and floating cells were harvested and fixed in 70% (vol/vol) ethanol. The cells were then stained with propidium iodide for 20 min in the dark. At least 1 × 105 stained cells were analysed for Sub-G1 profile on a FACScan Flow Cytometer (Becton Dickinson, San Jose, CA, USA). The percentage of sub-G1 (apoptotic) cells in mock-infected cells and infected cells was calculated. The results presented are averages and standard deviations from three separate experiments.
Western blot analysis
In order to compare transactivation function among Ad-p53 14/19, Ad-p53 22/23 and Ad-wt p53, levels of p53, Mdm2, p21WAF-1 and Bax were examined in SJSA and HT1080 cells (1.5 × 106 cells/10-cm dish) infected by NCV, Ad-wt p53, Ad-p53 14/19 and Ad-p53 22/23. To analyse the levels of phosphorylated p53, phosphorylated Mdm2 and Bax, SJSA and HT1080 cells were plated with 1.5 × 106 cells/10-cm dish. Cells were infected with Ad-wt p53 or Ad-p53 14/19 and treated with doxorubicin as described above. Cells were lysed in RIPA buffer (50 mM Tris-HCl, 1% NP40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM sodium fluoride). 100 μg of protein from cell lysates was separated by SDS–PAGE. Western blots were stained with antibodies against p53, Mdm2 (both kindly provided by Arnold Levine), phosphorylated p53 (Ser-6, -9, -15, -20, -37 and -46, Cell Signaling Technology, Beverly, MA, USA), phosphorylated Mdm2 (Ser-166, Cell Signaling Technology, Beverly, MA, USA) and Bax (Transduction Laboratories, Lexington, KY, USA). In order to observe the expression of multidrug-resistance proteins in NHF, HT1080 and SJSA, the MDR, MRP1 and MRP4 antibodies (Santa Cruz Biotechnology, Inc., CA, USA) were used. Protein levels were standardized with a monoclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; Chemicon International, Inc., Temecula, CA, USA). Blots were scanned with Image Quant software to detect proteins using an electrochemifluorescence detection system (Amersham Corp., Arlington Heights, IL, USA) on a Molecular Dynamics Storm PhosphorImager (Sunnyvale, CA, USA).
Results
Induction of apoptosis and p53 downstream targets by adenovirus-p53 vectors
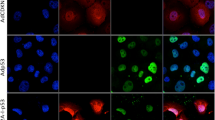
To determine whether p53 14/19 and p53 22/23 could induce apoptosis, two human sarcoma cell lines with either low (HT1080) or high (SJSA) levels of Mdm2 were utilized (Figure 1D). Normal human skin fibroblasts with low levels of Mdm2 were used to assess the toxicity of Ad-p53 14/19 and Ad-p53 22/23. As shown in Figure 1, apoptosis was significantly induced by Ad-wt p53 and Ad-p53 14/19 and to a lesser degree by Ad-p53 22/23 in low Mdm2-expressing HT1080 cells. Apoptotic cells increased seven- to eight-fold after infection with Ad-wt p53 or Ad-p53 14/19 and four-fold after infection with Ad-p53 22/23, when compared to uninfected cells or cells infected with NCV (Figure 1A). In contrast, in high Mdm2 expressing SJSA cells, there was a marked difference between Ad-wt p53 and Ad-p53 14/19. There was a four-fold increase in apoptotic cells in SJSA infected with Ad-p53 14/19 compared to Ad-wt p53, and a two-fold increase of apoptosis in SJSA infected with Ad-p53 22/23 compared to Ad-wt p53 (Figure 1B). No induction of apoptosis was detected in normal fibroblasts after infection by any adenoviral construct (Figure 1C). These results indicate that Ad-p53 14/19 and Ad-p53 22/23, both defective in Mdm2 binding, can induce apoptosis in HT1080 and SJSA cells regardless of Mdm2 expression, but have minimal effect on normal cells. Ad-p53 14/19 induces significant apoptosis in SJSA cells despite the overexpression of Mdm2.

Ad-p53 14/19 induces significant apoptosis in sarcoma cell lines. (A) HT1080 fibrosarcoma cells, (B) SJSA osteosarcoma cells and (C) NHF were infected with NCV or Ad-wt p53, Ad-p53 14/19 or Ad-p53 22/23. Cells were stained with propidium iodide (PI), apoptosis was assessed with sub-G1 profile analysis using a FACScan flow cytometer and fold increase of apoptotic cells was calculated. Values shown are the mean+standard deviation of Log [PI] from three independent experiments. * Indicates a significant increase compare to Ad-wt p53 treatment (P<0.05 in a two-tail Student's t-test). (D) The expression of Mdm2 in SJSA, HT1080 and NHF. Proteins were extracted from untreated cells and analysed by Western blot using antibodies against Mdm2. GAPDH is the protein loading control.
In Figure 2, it is apparent that p53 14/19 and p53 22/23 have different transcriptional activation activity. The 22/23 mutant has lost its transcriptional activation activity, but the 14/19 mutant retains the activity of the wild-type protein (Lin et al, 1994). Ad-p53 14/19 induced the expression of p53 target proteins Mdm2, p21WAF-1and Bax in both HT1080 and SJSA, and in SJSA there was greater induction of Mdm2 and p21WAF-1 than with Ad-wt p53. Ad-p53 22/23 had the same ability as Ad-p53 14/19 to induce Bax, but failed to induce Mdm2 and p21WAF-1 in either cell line. Since p53 14/19 retains its transactivation capability and induces greater apoptosis than p53 22/23 in both HT1080 and SJSA cells, we selected p53 14/19 for further investigation to determine if it could sensitize cells to apoptosis caused by doxorubicin or cisplatin.

Ad-p53 14/19 is a more potent inducer than Ad-p53 22/23 for p21WAF-1 and Mdm2, but not Bax in HT1080 and SJSA cell lines. Both cell lines were infected with NCV, Ad-wt p53, Ad-p53 22/23 and Ad-p53 14/19. After 24 h, whole-cell proteins were extracted and analysed by Western blot using antibodies against p53, Mdm2, p21WAF-1 and Bax, respectively. GAPDH is the protein loading control.
Adenovirus-p53 14/19 sensitizes sarcoma cells to doxorubicin and cisplatin
We analysed whether Ad-p53 14/19 could sensitize sarcoma cells to apoptosis induced by doxorubicin regardless of Mdm2 expression level. As shown in Figures 3 and 4, HT1080 is relatively sensitive and SJSA relatively resistant to doxorubicin treatment. After exposure to 0.1 μg ml−1 of doxorubicin, 35% of HT1080 cells were apoptotic (Figure 3B), while only 4% of SJSA cells were apoptotic (Figure 4B). When doxorubicin was combined with Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19, 66–83% of HT1080 cells underwent apoptosis (Figure 3B). However, in SJSA cells, when doxorubicin was combined with Ad-wt p53, only 15% of cells were apoptotic (Figure 4B), demonstrating continued resistance. In contrast, doxorubicin plus Ad-p53 22/23 or Ad-p53 14/19 generated greater apoptosis (44 or 63%) in SJSA cells. This demonstrates that Ad-p53 22/23 and Ad-p53 14/19 can not only enhance apoptosis induced by doxorubicin in sensitive cells such as HT1080 but can also overcome Mdm2-mediated doxorubicin resistance, as in SJSA cells. Moreover, the results suggest that Ad-p53 14/19 has more significant synergy with doxorubicin than Ad-p53 22/23.

Ad-p53 14/19, Ad-p53 22/23 and Ad-wt p53 enhance doxorubicin (Doxo)-mediated apoptosis in HT1080 fibrosarcoma cells. Cells were infected with NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 for 3 days alone or in combination with 0.1 μg/ml doxorubicin. Cells were then stained with PI and apoptosis was assessed with sub-G1 profile analysis using a FACScan flow cytometer. Examples of apoptosis are shown in (A). (B) Percentages of apoptotis are given as the mean+standard deviation of Log [PI] from three independent experiments. Gray bars indicate HT1080 cells treated by mock infection, doxorubicin, NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 alone. Black bars indicate cells treated with doxorubicin plus NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19.

Ad-p53 14/19 enhances doxorubicin-mediated apoptosis in SJSA osteosarcoma cells. Cells were infected with NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 for 3 days alone or in combination with 0.1 μg ml−1 doxorubicin. Cells were then stained with PI and apoptosis was assessed with sub-G1 profile analysis using a FACScan flow cytometer. Examples of apoptosis in SJSA cells are shown in (A). (B) Percentages of apoptotic cells are given as the mean+standard deviation of Log [PI] from three independent experiments. * Indicates a significant increase compared to Ad-wt p53 plus doxorubicin treatment (P<0.001 in a two-tail Student's t-test). Gray bars indicate SJSA cells treated by mock infection, doxorubicin, NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 alone. Black bars indicate cells treated with doxorubicin plus NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19.
In order to further demonstrate that Ad-p53 14/19 can increase apoptosis induced by a chemotherapeutic agent, cisplatin was selected. Cisplatin is a common chemotherapeutic agent for sarcomas, such as osteosarcoma (Stine et al, 2003). As shown in Figure 5, HT1080 is still relatively more sensitive to cisplatin treatment than SJSA. After exposure to 5 μg ml−1 of cisplatin, 70% of HT1080 cells were apoptotic (Figure 5A), while only 24% of SJSA cells were apoptotic (Figure 5B). When cisplatin was combined with Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19, around 84–88% of HT1080 cells underwent apoptosis (Figure 5A). However, in SJSA cells, when cisplatin was combined with Ad-wt p53 and Ad-p53 22/23, only 33–39% of cells were apoptotic (Figure 5B). In contrast, cisplatin plus Ad-p53 14/19 generated greater apoptosis (80%) in SJSA cells. This demonstrates that Ad-p53 14/19 can not only enhance apoptosis induced by doxorubicin but can also enhance apoptosis induced by cisplatin in either HT1080 or SJSA cells.

Ad-p53 14/19 combined with cisplatin induces more significant apoptosis than Ad-wt p53 combined with cisplatin in SJSA cells. Cells were infected with NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 for 3 days alone or in combination with 5 μg/ml cisplatin. Cells were then stained with PI and apoptosis was assessed with sub-G1 profile analysis using a FACScan flow cytometer. Percentages of apoptosis in HT1080 cells are shown in (A), and percentages of apoptosis in SJSA cells are shown in (B). Values shown are the mean+standard deviation of Log [PI] from three independent experiments. * Indicates a significant increase compared to Ad-wt p53 plus cisplatin treatment (P<0.005 in a two-tail Student's t-test). Gray bars indicate SJSA cells treated by mock infection, cisplatin, NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19 alone. Black bars indicate cells treated with cisplatin plus NCV, Ad-wt p53, Ad-p53 22/23 or Ad-p53 14/19.
Doxorubicin induces phosphorylation of wt p53 and p53 14/19 proteins
To evaluate phosphorylation of p53 and expression of its target proteins, SJSA and HT1080 were infected with Ad-wt p53 or Ad-p53 14/19 in conjunction with exposure to 0.1 μg ml−1 doxorubicin. Western blotting showed that expression of p53 was induced in cells treated with both Ad-wt p53 plus and Ad-p53 14/19 plus doxorubicin (Figure 6). In SJSA cells, Ad-p53 14/19 alone resulted in a higher p53 expression and greater induction of downstream targets Mdm2 and Bax than Ad-wt p53. In HT1080 cells, both Ad-p53 14/19 and Ad-wt p53 induced p53 target proteins Mdm2 and Bax. Mdm2 was phosphorylated in cells infected with Ad-p53 14/19 and Ad-wt p53 alone.

Doxorubicin induces phosphorylation of p53 in HT1080 (low Mdm2) and SJSA (high Mdm2). Cells were treated with Ad-p53 or Ad-p53 14/19 and 0.1 μg/ml doxorubicin (Doxo). After 24 h, whole-cell proteins were extracted and analysed by Western blot using antibodies against p53, phosphorylated p53 (Ser-6, Ser-15 Ser-20, Ser-37 and Ser-46), Mdm2, phosphorylated Mdm2 (Ser-166) and Bax. GAPDH is the protein loading control.
In both HT1080 and SJSA cells, there was slightly higher p53 expression in cells treated with Ad-p53 14/19 plus doxorubicin than in cells treated with Ad-wt p53 plus doxorubicin (Figure 6). This suggests that doxorubicin stabilizes wt p53 protein and may further stabilize p53 14/19. In both cell lines treated with Ad-wt p53 plus doxorubicin or Ad-p53 14/19 plus doxorubicin, there were dramatic increases in p53 phosphorylation at Ser-6, Ser-15, Ser-20, Ser-37 and Ser-46. p53 phosphorylation mediated by doxorubicin may enhance Bax (Figure 6) and p21WAF-1 (data not shown) induction in SJSA cells. This serine phosphorylation is weakly or not detected in cells infected with Ad-wt p53 or Ad-p53 14/19 alone or cells treated with doxorubicin alone. In SJSA cells, Mdm2 was slightly increased in Ad-wt p53-infected cells, but was dramatically increased in Ad-p53 14/19-infected cells. In both cell lines, Mdm2 expression and phosphorylation were decreased in cells treated with doxorubicin alone or Ad-wt p53 plus doxorubicin, and markedly diminished in cells infected with Ad-p53 14/19 plus doxorubicin (Figure 6).
Discussion
Derangements of the p53 pathway are common in soft-tissue sarcomas and osteosarcomas. Mdm2 overexpression occurs frequently, and these tumours typically contain wt p53 (Oliner et al, 1992). Since Mdm2 inactivates and promotes degradation of p53, the ability to restore p53 function by introduction of wt p53 into sarcomas is limited. We tested two modified p53 genes that are not responsive to the Mdm2 autoregulatory feedback loop (Lin et al, 1994). Ad-p53 22/23, an adenovirus vector encoding a transcription-defective p53 mutant, is able to induce limited apoptosis compared to Ad-wt p53 in both sarcoma cell lines tested, even combined with doxorubicin or cisplatin. Since both cell lines express endogenous wt p53 (Evdokiou and Cowled, 1998), it is possible that p53 22/23 may form dimers or tetramers with endogenous wt p53. These hetero-tetramers may retain partial function as a transcription factor. Interestingly, Ad-p53 22/23, although unable to induce Mdm2 and p21WAF-1, can still induce Bax expression in these cell lines. Bax is one of the p53 primary-response genes involved in the induction of apoptosis (Miyashita and Reed, 1995); this may explain in part why Ad-p53 22/23 is able to induce limited apoptosis in these cell lines. In addition to induction of apoptosis by transcription-dependent mechanisms, transcription-independent mechanism(s) have been reported for p53 (Haupt et al, 1997b; Yan et al, 1997; Mihara et al, 2003). We observed that Ad-p53 22/23 failed to induce apoptosis in the Caov-3 cell line, but induced limited apoptosis in MDAH 2774 cell line (data not shown). Both ovarian cancer cell lines harbour p53 mutations (Yaginuma and Westphal, 1992). Therefore, p53 22/23 may still be able to induce apoptosis via a transcription-independent mechanism(s).
p53 14/19 maintains its transcriptional activation and antiproliferative functions in the face of high Mdm2 levels (Chen et al, 1995, 1996). Our data indicate that p53 14/19 has greater ability to activate Mdm2 and p21WAF-1 than p53 22/23, and induces more significant apoptosis than p53 22/23 in sarcoma cells. These results suggest that p53 14/19 would be the better construct to use in further research targeting sarcoma cell lines with high levels of Mdm2. In addition, p53 14/19 more dramatically induced p21WAF-1 than wt p53 in SJSA cells, but there was equivalent induction of p21WAF-1 in HT1080 cells. There appears to be no relationship between the induced level of p21WAF-1 and the induced apoptosis by wt p53 or p53 14/19 in HT1080 or SJSA, suggesting that the induction of p21WAF-1 is not responsible for p53-dependent apoptosis in this model system. The major function of p21WAF-1 is to induce cell cycle growth arrest at the G1 phase (Medrano et al, 1995). Therefore, it is likely that other downstream targets of p53 are more directly involved with induction of apoptosis induced by p53 14/19.
We have shown that Ad-p53 14/19 induces dramatic apoptosis in sarcoma cell lines with either low or high Mdm2 expression. In HT1080 cells (low Mdm2), Ad-p53 14/19 induced apoptosis similar to Ad-wt p53. In SJSA cells (high Mdm2), Ad-p53 14/19 induced dramatically more apoptosis than Ad-wt p53 (44 vs 11%). These results are consistent with previous Ad-p53 14/19 data from cell growth inhibition assays (Lin et al, 2000). We also observed that Ad-p53 14/19 more potently inhibits SJSA cells’ growth than Ad-wt p53 in nude mice (Tang and Lin, unpublished data). Neither construct appeared to be toxic to normal skin fibroblasts in vitro. Two possible explanations for this selectivity are that cancer cells may be more sensitive to adenoviral transfer of wt p53, p53 22/23 or p53 14/19, or that oncogenes such as c-myc and cyclin D1 are overexpressed in malignant but not normal cells. Overexpression of these oncogenes sensitizes cells to apoptosis induced by wt p53 (Hermeking and Eick, 1994; Wagner et al, 1994; Lin et al, 1996; Hiyama and Reeves, 1999). We observed no evidence that adenoviral transfer of p53 14/19 would be any less safe than transfer of wt p53 to normal cells.
Chemoresistance is a major problem in the treatment of malignant tumours; hence, it will be of potential value to discover combination protocols that overcome resistance. Doxorubicin, a topoisomerase II inhibitor, is commonly used in the treatment of sarcomas (Sondak and Chang, 2001). Doxorubicin causes apoptosis by direct DNA damage, at least in part in a p53-dependent manner (Lowe et al, 1993, 1994; Malcomson et al, 1995; Vikhanskaya et al, 1997; Weinstein et al, 1997; Blagosklonny and El-Deiry, 1998). Cisplatin is also one of the most widely used chemotherapeutic agents for the treatment of many types of cancer. Apoptosis is the primary mode of cell death induced by cisplatin, and cisplatin occurs primarily through its ability to bind covalently to DNA and prevent DNA replication and transcription (Li et al, 1998; Masuda et al, 1998). The HT1080 cell line is sensitive to doxorubicin (Li et al, 2001), and the present study shows that HT1080 is also more sensitive to cisplatin. We found that SJSA was resistant to doxorubicin and not more sensitive to cisplatin than HT1080. These results are in agreement with one previous report that the mdm2 gene caused resistance to doxorubicin but not to cisplatin in some sarcoma cell lines, and in these lines there was an increase in the expression of the mdr-1 gene that encodes P-gp (Cocker et al, 2001). However, we did not observe the expression of multidrug resistance proteins, such as MDR, MRP1 and MRP4 in NHF, HT1080 and SJSA cell lines (data not shown). Our data suggest that Ad-p53 14/19, but not Ad-wt p53 is able to overcome this resistance to doxorubicin as well as enhance SJSA cells’ sensitivity to cisplatin. Nevertheless, Ad-p53 22/23 is also able to increase apoptosis induced by doxorubicin in SJSA cells’ sensitize has the same effect as Ad-wt p53 in enhancing apoptosis induced by cisplatin (Figure 4B and 5B). Ad-p53 14/19 and Ad-p53 22/23 also augmented the apoptotic response to doxorubicin and cisplatin in HT1080, the sensitive cell line (Figure 3B and 5A).
p53 phosphorylation is induced by DNA damage at a variety of sites including Ser-6, Ser-15, Ser-20, Ser-37 and Ser-46 (Sakaguchi et al, 1998; Tibbetts et al, 1999; Higashimoto et al, 2000; Oda et al, 2000; Achanta et al, 2001; Shono et al, 2002). These modifications may stabilize and activate p53 as a transcription factor. (Shieh et al, 1997; Siliciano et al, 1997; Chehab et al, 2000). Our studies show that both Ad-wt p53 and Ad-p53 14/19 in combination with doxorubicin dramatically increase p53 phosphorylation. Previous studies have shown that phosphorylation at Ser-15, Ser-20 and Ser-46 is critical for regulating apoptotic activity (Oda et al, 2000; Shono et al, 2002). Phosphorylation of Ser-15, Ser-20 and Ser-37 interferes with Mdm2 interaction and impairs the ability of Mdm2 to inhibit p53-dependent transactivation (Shieh et al, 1997; Chehab et al, 1999; Appella and Anderson, 2000). We found that both wt p53 and p53 14/19 are phosphorylated at Ser-6, Ser-15 Ser-20, Ser-37 and Ser-46 in response to doxorubicin. Hence, the mutations at residues 14 and 19 do not affect these phosphorylation sites. Interestingly, levels of Ser-6 and Ser-46 phosphorylation of p53 14/19 were much higher than wt p53 in SJSA cells.
Infection with Ad-wt p53 combined with doxorubicin resulted in enhanced apoptosis compared to Ad-wt p53 or doxorubicin alone in HT1080 but not in SJSA cells. This suggests that negative regulation of p53 by Mdm2 in SJSA cells may limit the magnitude of p53 activation and p53-dependent apoptosis even when p53 is phosphorylated. Mdm2 overexpression diminished the apoptotic response to doxorubicin even when exogenous wild-type p53 was reintroduced, consistent with other reports (Cocker et al, 2001).
Doxorubicin treatment reduced Mdm2 expression and phosphorylation in both cell lines. This is consistent with a previous report that Mdm2 is downregulated by anticancer drugs including doxorubicin (Gao et al, 1999). Doxorubicin can activate p53 in Mdm2-overexpressing cells by decreasing Mdm2 levels, alleviating the inhibition of p53.
We observed increased expression of Bax in SJSA cells treated with Ad-wt p53 plus doxorubicin compared to cells infected with Ad-wt p53 alone. Bax levels correlated with p53-induced apoptosis. In HT1080 cells, although the expression of Bax was induced by Ad-wt p53 or Ad-p53 14/19, there was no relationship between Bax expression and apoptosis. Previous studies have shown that Bax induction by p53 is necessary to inhibit tumour growth (Yin et al, 1997), but that the contribution of Bax to p53-mediated apoptosis is cell-type dependent (Knudson et al, 1995; McCurrach et al, 1997). In HT1080 cells, other downstream targets of p53 may be more important for induction of apoptosis.
In conclusion, Mdm2 overexpression can promote cancer cell resistance to DNA-damaging agents and limit the effectiveness of chemotherapeutic drugs. Modified p53, particularly p53 14/19, retains the proapoptotic and transcriptional activity of wt p53, and can augment the effectiveness of chemotherapy even in cells overexpressing Mdm2. Strategies involving p53 14/19 with chemotherapeutic agents may be a useful approach for sarcomas and other tumours with high levels of Mdm2.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Achanta G, Pelicano H, Feng L, Plunkett W, Huang P (2001) Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res 61: 8723–8729
Appella E, Anderson C (2000) Signaling to p53: breaking the posttranslational modification code. Pathol Biol 48: 227–245
Beaudry G, Bertelsen A, Sherman M (1996) Therapeutic targeting of the p53 tumor suppressor gene. Curr Opin Biotechnol 7: 592–600
Blagosklonny M, El-Deiry W (1998) Acute overexpression of wt p53 facilitates anticancer drug-induced death of cancer and normal cells. Int J Cancer 75: 933–940
Chan H, Thorner P, Haddad G, Ling V (1990) Immunohistochemical detection of P-glycoprotein: prognostic correlation in soft tissue sarcoma of childhood. J Clin Oncol 8: 689–704
Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 9613777-82: 13777–13782
Chehab N, Malikzay A, Appel M, Halazonetis T (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev 14: 278–288
Chen J, Lin J, Levine A (1995) Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol Med 1: 142–152
Chen J, Wu X, Lin J, Levine A (1996) mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol 16: 2445–2452
Cocker H, Hobbs S, Tiffin N, Pritchard-Jones K, Pinkerton C, Kelland L (2001) High levels of the MDM2 oncogene in paediatric rhabdomyosarcoma cell lines may confer multidrug resistance. Br J Cancer 85: 1746–1752
Donehower LA, Bradley A (1993) The tumor suppressor p53. Biochim Biophys 1155: 181–205
Evdokiou A, Cowled PA (1998) Tumor-suppressive activity of the growth arrest-specific gene GAS1 in human tumor cell lines. Int J Cancer 75: 568–577
Gao C, Nakajima T, Taya Y, Tsuchida N (1999) Activation of p53 in MDM2-overexpressing cells through phosphorylation. Biochem Biophys Res Commun 2: 860–864
Hann B, Balmain A (2003) Replication of an E1B 55-kilodalton protein-deficient adenovirus (ONYX-015) is restored by gain-of-function rather than loss-of-mutants. J Virol 77: 11588–11595
Haupt Y, Maya R, Kazaz A, Oren M (1997a) mdm2 promotes the rapid degradation of p53. Nature 387: 296–299
Haupt Y, Rowan S, Shaulian E, Kazaz A, Vousden K, Oren M (1997b) p53 mediated apoptosis in HeLa cells: transcription dependent and independent mechanisms. Leukemia 13: 337–339
Hermeking H, Eick D (1994) Mediation of c-Myc-induced apoptosis by p53. Science 265: 2091–2093
Higashimoto Y, Saito S, Tong XH, Hong A, Sakaguchi K, Appella E, Anderson C (2000) Human p53 is phosphorylated on serines 6 and 9 in response to DNA damage-inducing agents. J Biol Chem 275: 23199–23203
Hiyama H, Reeves S (1999) Role for cyclin D1 in UVC-induced and p53-mediated apoptosis. Cell Death Differ 6: 565–569
Hoffman J, Schmidt-Peter P, Hansch W, Naundorf H, Bunge A, Becker M, Fichtner I (1999) Anticancer sensitivity and expression of multidrug resistance markers in early passage human sarcomas. Clin Cancer Res 5: 2198–2204
Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27
Jiminez R, Zalupski M, Frank J, Du W, Ryan J, Lucas D (1999) Multidrug resistance phenotype in high grade soft tissue sarcoma: correlation of P-glycoprotein immunohistochemistry with pathologic response to chemotherapy. Cancer 86: 976–981
Joensuu H, Roberts P, Sarlomo-Rikala M, Andersson L, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri G (2001) Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 344: 1052–1056
Keleti J, Quezado M, Abaza M, Raffeld M, Tsokos M (1996) The MDM2 oncoprotein is overexpressed in rhabdomyosarcoma cell lines and stabilizes wild-type p53 protein. Am J Pathol 149: 143–151
Knudson C, Tung K, Tourtellotte W, Brown G, Korsmeyer S (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99
Ko S, Gotoh A, Thalmann G, Zhau H, Johnston D, Zhang W, Kao C, Chung L (1996) Molecular therapy with recombinant p53 adenovirus in an androgen-independent, metastatic human prostate cancer model. Hum Gene Ther 7: 1683–1691
Kondo S, Barnett G, Hara H, Morimura T, Takeuchi J (1995) MDM2 protein confers the resistance of a human glioblastoma cell line to cisplatin-induced apoptosis. Oncogene 10: 2001–2006
Kondo S, Kondo Y, Hara H, Kaakaji R, Peterson J, Morimura T, Takeuchi J, Barnett G (1996) Mdm2 gene mediates the expression of mdr1 gene and P-glycoprotein in a human glioblastoma cell line. Br J Cancer 74: 1263–1268
Kubbutat M, Jones S, Vousden K (1997) Regulation of p53 stability by mdm2. Nature 387: 299–303
Kubbutat M, Ludwig R, Ashcroft M, Vousden K (1998) Regulation of mdm2-directed degradation by the C terminus of p53. Mol Cell Biol 18: 5690–5698
Landers J, Cassel S, George D (1997) Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res 57: 3562–3568
Leach F, Tokino T, Meltzer P, Burrell M, Oliner J, Smith S, Hill D, Sidransky D, Kinzler K, Vogelstein B (1993) p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res 53: 2231–2234
Levine AJ, Momand J, Finlay CA (1991) The p53 tumour suppressor gene. Nature 351: 453–456
Li Q, Gardner K, Zhang L, Tsang B, Bostick-Bruton F, Reed E (1998) Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J Biol Chem 273: 23419–23425
Li W, Takahashi N, Jhanwar S, Cordon-Cardo C, Elisseyeff Y, Jimeno J, Faircloth G, Bertino J (2001) Sensitivity of soft tissue sarcoma cell lines to chemotherapeutic agents: identification of ecteinascidin-743 as a potent cytotoxic agent. Clin Cancer Res 7: 2908–2911
Lin J, Chen J, Elenbaas B, Levine AJ (1994) Several hydrophobic amino acids in the p53 N-terminal domain are required for transcriptional activation, binding to mdm-2 and the Adenovirus 5 E1B 55kd protein. Genes Dev. 8: 1235–1246
Lin J, Jin X, Page C, Sondak VK, Jiang G, Reynolds KR (2000) A modified p53 overcomes mdm2-mediated oncogenic transformation. Cancer Res 60: 5895–5901
Lin J, Reichner C, Wu X, Levine AJ (1996) Analysis of wild-type and mutant p21WAF-1 gene activities. Mol Cell Biol 16: 1786–1793
Lowe S, Bodis S, McClatchey A, Remington L, Ruley H, Fisher D, Housman D, Jacks T (1994) p53 status and the efficacy of cancer therapy in vivo. Science 266: 807–810
Lowe S, Ruley H, Jacks T, Housman D (1993) p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74: 957–967
Malcomson R, Oren M, Wyllie A, Harrison D (1995) p53-independent death and p53-induced protection against apoptosis in fibroblasts treated with chemotherapeutic drugs. Br J Cancer 72: 952–957
Masuda H, Ozols R, Lai G, Fojo A, Rothenberg M, Hamilton T (1998) Increased DNA repair as a mechanism of acquired resistance to cis-diamminedichloroplatinum (II) in human ovarian cancer cell lines. Cancer Res 48: 5713–5716
McCurrach M, Connor T, Knudson C, Korsmeyer S, Lowe S (1997) bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci USA 94: 2345–2349
Medrano EE, Im S, Yang F, Abdel-Malek ZA (1995) Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res 55: 4047–4052
Meng R, Shih H, Prabhu N, George D, el-Deiry W (1998) Bypass of abnormal MDM2 inhibition of p53-dependent growth suppression. Clin Cancer Res 4: 251–259
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll U (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590
Miyashita T, Reed J (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299
Momand J, Jung D, Wilczynski S, Niland J (1998) The MDM2 gene amplification database. Nucleic Acids Res 26: 3453–3459
Momand J, Zambetti G, Olson D, George D, Levine A (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69: 1237–1245
Nakayama T, Toguchida J, Wadayama B, Kanoe H, Kotoura Y, Sasaki M (1995) MDM2 gene amplification in bone and soft-tissue tumors: association with tumor progression in differentiated adipose-tissue tumors. Int J Cancer 64: 342–346
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862
Oliner J, Kinzler K, Meltzer P, George D, Vogelstein B (1992) Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358: 80–83
Oliner J, Pietenpol J, Thiagalingam S, Gyuris J, Kinzler K, Vogelstein B (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362: 857–860
Prives C, Hall P (1999) The p53 pathway. J Pathol 187: 112–126
Sakaguchi K, Herrera J, Saito S, Miki T, Bustin M, Vassilev A, Anderson C, Appella E (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev 15: 2831–2841
Shieh S, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334
Shono T, Tofilon PJ, Schaefer TS, Parikh D, Liu TJ, Lang FF (2002) Apoptosis induced by adenovirus-mediated p53 gene transfer in human glioma correlates with site-specific phosphorylation. Cancer Res 62: 1069–1076
Siliciano J, Canman C, Taya Y, Sakaguchi K, Appella E, Kastan M (1997) DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev 11: 3471–3481
Sondak V, Chang A (2001) Clinical evaluation and treatment of soft tissue tumors. In: Weiss SW, Goldblum JR (eds) Enzinger and Weiss's Soft Tissue Tumors, 4th edn, pp. 21–44. Missouri: Mosby, St Louis
Stine K, Saylors R, Saccente S, Becton DL (2003) Long-term survival in osteosarcoma patients following retinoblastoma using doxorubicin, cisplatin, and methotrexate. Med Pediatr Oncol 41: 77–78
Suzuki A, Toi M, Yamamoto Y, Saji S, Muta M, Tominaga T (1998) Role of MDM2 overexpression in doxorubicin resistance of breast carcinoma. Jpn J Cancer Res 89: 221–227
Tibbetts R, Brumbaugh K, Williams J, Sarkaria J, Cliby W, Shieh S, Taya Y, Prives C, Abraham R (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 15: 152–157
Vikhanskaya F, Clerico L, Valenti M, Stanzione M, Broggini M, Parodi S, Russo P (1997) Mechanism of resistance to cisplatin in a human ovarian-carcinoma cell line selected for resistance to doxorubicin: possible role of p53. Int J Cancer 72: 155–159
Vogelstein B, Kinzler KW (1992) p53 function and dysfunction. 70: 523–526
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310
Wagner A, Kokontis J, Hay N (1994) Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev 8: 2817–2830
Weinstein J, Myers T, O'Connor P, Friend S, Fornace AJ, Kohn K, Fojo T, Bates S, Rubinstein L, Anderson N, Buolamwini J, van Osdol W, Monks A, Scudiero D, Sausville E, Zaharevitz D, Bunow B, Viswanadhan V, Johnson G, Wittes R, Paull K (1997) An information-intensive approach to the molecular pharmacology of cancer. Science 275: 343–349
Yaginuma Y, Westphal H (1992) Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res 52: 4196–4199
Yan Y, Shay J, Wright W, Mumby M (1997) Inhibition of protein phosphatase activity induces p53-dependent apoptosis in the absence of p53 transactivation. J Biol Chem 2772: 15220–15226
Yang C, Cirielli C, Capogrossi M, Passaniti A (1995) Adenovirus-mediated wild-type p53 expression induces apoptosis and suppresses tumorigenesis of prostatic tumor cells. Cancer Res 55: 4210–4213
Yin C, Knudson C, Korsmeyer S, Van DT (1997) Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385: 637–640
Zhan M, Yu D, Lang A, Li L, Pollock R (2001) Wild type p53 sensitizes soft tissue sarcoma cells to doxorubicin by down-regulating multidrug resistance-1 expression. Cancer 92: 1556–1566
Zhang W, Alemany R, Wang J, Koch P, Ordonez N, Roth J (1995) Safety evaluation of Ad5CMV-p53 in vitro and in vivo. Hum Gene Ther 6: 155–164
Acknowledgements
We thank Mats Ljungman at the University of Michigan Comprehensive Cancer Center for providing normal human skin fibroblasts, and Dana Gossett for careful reading of this manuscript. This work was supported in part by the Michigan Life Sciences Corridor grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tang, HJ., Qian, D., Sondak, V. et al. A modified p53 enhances apoptosis in sarcoma cell lines mediated by doxorubicin. Br J Cancer 90, 1285–1292 (2004). https://doi.org/10.1038/sj.bjc.6601653
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601653
Keywords
This article is cited by
-
Adenovirus-mediated p53 tumor suppressor gene therapy of osteosarcoma
Laboratory Investigation (2006)
-
Response rate of fibrosarcoma cells to cytotoxic drugs on the expression level correlates to the therapeutic response rate of fibrosarcomas and is mediated by regulation of apoptotic pathways
BMC Cancer (2005)
-
Mdm2 and mdmX prevent ASPP1 and ASPP2 from stimulating p53 without targeting p53 for degradation
Oncogene (2005)
