
Abstract
Mutations in the GLRA1 gene, which encodes the α1-subunit of the inhibitory glycine receptor (GlyR), are the underlying causes in the majority of cases of hereditary startle disease (OMIM no. 149400). GlyRs are modulated by alcohols and volatile anesthetics, where a specific amino acid at position 267 has been implicated in receptor modulation. We describe a hyperekplexia family carrying the novel dominant missense allele GLRA1(S267N), that affects agonist responses and ethanol modulation of the mutant receptor. This study implies that a disease-related receptor allele carries the potential to alter drug responses in affected patients.
Similar content being viewed by others

Introduction
Strychnine-sensitive glycine receptors (GlyRs) mediate fast synaptic inhibition in mammalian spinal cord and brainstem. GlyRs belong to the nicotinic acetylcholine receptor superfamily of ligand-gated ion channels. The hypertonic motor disorder hyperekplexia (STHE, OMIM no. 149400),1 is caused primarily by mutant alleles of the glycine receptor α1 gene (GLRA1; NM_000171.2), while alterations of the glycine transporter GLYT2 (NM_004211.3) have also been observed.2, 3, 4 In the neonate, the disease manifests itself as stiff baby syndrome, while exaggerated startle responses predominate in the adult. Dominant forms of STHE have been attributed to mutations within the pore-lining transmembrane segment TM2 and adjacent regions, while recessive cases result from mutations within TM15 and from a null allele of GLRA1.6 GlyRs are molecular targets for alcohols and anesthetics7 where modulation was attributed to a serine residue at position α1(267).7, 8 Here, we report the novel allele GLRA1(S267N), associated with dominant hyperekplexia. Electrophysiological and biochemical properties of the mutant channels were examined in HEK293 cells.
Patients and methods
Patients
Following the guidelines of the Local Ethics Committee, informed consent was obtained from the family for electrophysiological studies and mutation analysis of the GLRA1 and GLRB genes. Symptoms of stiff baby syndrome in the index patient (30-3, age=5 weeks) were diagnosed clinically shortly after birth, while his father (30-1, age=28 years) displayed the hallmark features of hyperekplexia in an adult. The mother of the patient was unaffected. After birth, patient 30-3 suffered from severe muscular hypertonia and hyperreflexia. Upon neurophysiological examination, startle responses affecting muscles of the head, neck and upper arms could be elicited by acoustic stimuli: reflex latencies from the masseter, trapezius and sternocleidomastoid muscles were shortened (<22 ms) and followed by an abnormal blink reflex (M. orbicularis oculi: 23–26 ms). In contrast, the symptoms of the patient's father were less pronounced: exaggerated brain stem reflexes were found by neurophysiological testing, but no acoustic startle could be elicited. In both patients, multiple EEG recordings were not indicative of epilepsy. The father of the patient reported no subjective differences in ethanol sensitivity.
Molecular analysis
Polymorphism analysis
DNA was extracted from EDTA-blood using a standard protocol. For initial screening, MALDI-TOF MS-based genotyping of known GLRA1 polymorphisms was performed as described previously.9 Direct sequencing was performed using an Automated DNA Sequencer (ABI – Perkin Elmer, 377A).
Mutagenesis and expression
The mutation G1180A was introduced into a GLRA1 cDNA plasmid. HEK293 cells were transfected by CaPO4 and utilized after 48–72 h.
Electrophysiology recordings and data analysis
Whole cell currents were recorded at 22–24°C and a holding potential of −60 mV using a U-tube for rapid agonist delivery and an EPC 9 recording system (HEKA Electronics, Germany) as described previously.10 Extracellular solution contained (in mM): 137 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES; pH 7.4. Intracellular solution contained (in mM): 135 CsCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES, pH 7.4. Analysis was performed using Pulsefit (HEKA Electronics) and Origin software (OriginLab Corporation, USA). Concentration–response relationships were measured using at least 7 agonist concentrations (range=1–20 000 μ M) and fitted to the Hill equation. Data are shown as means±SEM.
[3H]strychnine binding
Specific binding was measured in triplicates by filtration. For displacement binding, membranes were incubated with 16.7 nM [3H]strychnine (GE Healthcare) and increasing concentrations of unlabeled ligands. For determination of ethanol dependence, membranes were incubated with increasing concentrations of [3H]strychnine (range=1–150 nM) in the presence or absence of 200 mM ethanol.
Results
Identification of GLRA1(S267N)
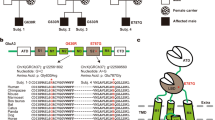
Genomic DNA was subjected to initial MALDI–TOF MS genotyping.9 After known mutations were excluded, GLRA1 amplimers were sequenced directly. Affected individuals were heterozygous for the substitution GLRA1(G1180A) in exon 7 (Figure 1a). This was confirmed by multiplex MALDI–TOF MS genotyping9 where the S267N allele was identified in the patient sample (Figure 1b). GLRA1(G1180A) predicts an asparagine instead of a serine residue at position 267 of GlyRα1. According to topology predictions, S267 locates to transmembrane domain 2 (TM2) close to the extracellular opening of the channel11 and is conserved between human and murine GlyRα-subunits.

Hyperekplexia allele of the GLRA1 gene in family FD. (a) Direct sequencing of exon 7 amplimers revealed heterozygosity for a nucleotide substitution G1180A in affected individuals (30-1, 30-3). On the reverse strand, this corresponds to C1180T. (b) Multiplex MALDI-TOF MS genotyping of the hyperekplexia alleles I244N (T1111A), S267N (G1180A) and K276E (A1206G). MALDI-TOF-MS spectra of the homozygous wild-type allele (control), and the heterozygous S267N allele (arrowhead, patient). Unextended primers (P-K276, 4489 Da; P-S267, 5398 Da; P-I244, 6364 Da) were used for internal mass calibration. For the S267N allele, the molecular mass of 6000 Da represented the wild-type G-allele (wt), while the molecular mass of 5695 Da indicated the mutated allele (G1180A). Alleles I244 (T1111A) and K276E (A1206G) were not present in the patient.
Electrophysiological studies
Constructs carrying the mutation GLRA1(G1180A) were transfected transiently into HEK293 cells. GlyR surface expression in membranes of α1S267N-transfected cells was unchanged from α1WT cells, as assessed by immunoblotting (data not shown). Whole-cell recordings showed no significant differences in maximum currents at between α1WT (Imax=6.3±1.7 nA) and α1S267N (Imax=5.4±1.7 nA, 3 mM glycine, n=10 each). Concentration–response analyses for glycine yielded a ≈17-fold reduction of the apparent affinity in α1S267N homomers compared to the wild type (Figure 2a). Apparent glycine affinity was reduced ∼2.3-fold for cells co-transfected with equimolar amounts of α1WT and α1S267N plasmid (Figure 2a). As synaptic GlyRs are thought to contain 2α1 and 3β subunits,12 α1WT, β and α1S267N constructs were expressed at ratios of 1/8 (α1WT/β) or 1/1/8 (α1WT/α1S267N/β). While α1WT/β heteromers displayed glycine responses comparable to α1WT homomers (data not shown), EC50 increased ∼5.5-fold for α1WT/α1S267N/β heteromers, consistent with incorporation of the mutant subunit (Figure 2a). While β-alanine and taurine are full agonists of wild-type GlyRs, both ligands act as either partial agonists or competitive antagonists in GlyRs with mutations in the intracellular or extracellular loops adjacent to TM2.13 Indeed, β-alanine and taurine showed a decreased efficacy on α1S267N GlyRs, eliciting only 17% (β-alanine, n=11), and 2% (taurine, n=8) of the maximum glycine response at 1 mM agonist (Figure 2b). Dose–response analysis for β-alanine on α1S267N GlyRs could be fitted under the assumption that KD and Imax were unchanged, and only channel opening was affected (Figure 2c), consistent with literature data.5

Agonist pharmacology of homo- and heteromeric α1WT and α1S267N GlyRs. (a) Dose–response curves. Peak currents were normalized to the response obtained with 3 mM glycine (black cross). Symbols are mean±SEM of normalized values (n=5 cells each, 3 measurements/concentration). Data were fitted to the Hill equation. α1S267N: EC50=720±24 μ M, nHill=3.3±0.3 (circles); α1WT: EC50=46±3 μ M, nHill=1.9±0.2 (black squares); α1S267N/α1WT: EC50=105±83 μ M, nHill=1.6±0.2 (triangles); α1WT/α1S267N/β: EC50=334±83 μ M, nHill=1.5±0.1 (open squares). Not shown: α1WT/β: EC50=60±13 μ M, nHill=1.5±0.1 (n=3). (b) 1 mM of glycine, β-alanine and taurine was applied to either α1WT or α1S267N. Currents were normalized to the response evoked by 1 mM glycine (left panel, α1WT: white bars, α1S267N: gray bars; sample traces right). (c) Dose–response relationship for β-alanine in α1S267N, currents normalized to 3 mM β-alanine. α1S267N: EC50=703±40 μ M; not shown α1WT: 120±15 μ M. The dose response relationship for glycine is displayed for comparison. Data were fitted using a 2-ligand model (R+2L<=>RL2<=>RL2(open)) and assuming identical KD, varying the channel-open equilibrium. (d) [3H]strychnine binding to membrane preparations of α1WT or α1S267N. Values were normalized to the [3H]Strychnine binding in the absence of competing ligand. α1WT: (in mM) Ki=0.183±0.033 (solid symbols); α1S265N Ki=2.3±0.22 (open symbols). For β-alanine and taurine, β-alanine: (in mM) Kiα1WT: 0.13±0.01; Kiα1S267N: 1.5±0.3; taurine: Kiα1WT: 0.2±0.02; Kiα1S267N: 1.65±0.09.
Radioligand displacement binding
[3H]strychnine displacement by GlyR agonists was determined to assess alterations in ligand binding. The average numbers of binding sites were not different (in pmol/g membranes; α1WT: Bmax=1.05, α1S267N: Bmax=0.82) and there was no significant change in [3H]strychnine affinity (in nM; KDα1WT=33.5±1.2; KDα1S267N=25.4±0.9), suggesting that antagonist binding was not changed in α1S267N GlyRs. Displacement by glycine, however, was shifted toward a higher Ki in α1S267N membranes Figure 2d), corresponding to a ≈15-fold reduced binding affinity. Binding affinities were diminished ≈21- and 15-fold for β-alanine and taurine, respectively, in α1S267N GlyRs (Figure 2d). The mutation α1S267N thus causes reduced agonist binding which is compatible with a reduced channel opening efficacy for partial agonists.
Ethanol modulation of α1S267N GlyRs
Ethanol sensitivity of recombinant GlyRs depends on both, the expression system used14 and the agonist concentration applied.15 Here, modulation of glycine-induced currents by alcohols was observed in ∼50% of cells at agonist concentrations of EC10–20. For α1WT, glycine responses were doubled following preincubation with 200 mM ethanol while potentiation of α1S267N was nearly absent (Figure 3a and b). Ethanol effects (200 mM) were quantified in [3H]strychnine binding assays with membranes carrying α1WT, α1S267N or α1S267Y, a mutation in which ethanol inhibited glycinergic currents.16 For α1WT and α1S267N, the apparent KD was shifted leftward with ethanol (Figure 3c and d). In contrast, the KD in α1S267Y was shifted rightward, consistent with inhibition by ethanol (Figure 3e). Ethanol modulation at 25 nM of [3H]strychnine (Figure 3f) revealed an almost two-fold increase of binding to α1WT membranes (P<0.01); no change was observed for α1S267N, and strychnine binding was reduced in α1S267Y membranes (P<0.05). Thus, the mutation α1S267N affects ethanol sensitivity of the glycine receptor α1-subunit.

Modulation of GlyR receptors by alcohols. (a) Whole cell responses to glycine were modulated by 200 mM ethanol in cells expressing α1WT (5 μ M glycine, EC10) or α1S267N (150 μ M glycine, EC20) receptors (representative traces shown, note the difference in scale). (b) Potentiation in comparison to control (%, no ethanol) is shown for n=10 cells expressing either α1WT (122±43) or α1S267N (5±13). (c–f) Equilibrium binding of [3H]strychnine to membranes of HEK293 cells transfected with α1WT or an α1S267-subunit variant was measured in the presence (open symbols) or absence (solid symbols) of 200 mM ethanol. Data are derived from triplicate measurements of three separate experiments each. A total of 16.7 nM [3H]strychnine (KD=11 nM) were used for experiments. Data were normalized to maximum binding of [3H]strychnine. α1WT (c, circles): (in nM) KD,_no_ETOH=35.6±3.1; KD,ETOH=19.2±3.6; α1S267N (d, squares): (in nM) KD,_no_ETOH=25.05±2.7; KD,ETOH=10.8±2.3; α1S267Y (e; diamonds): (in nM) KD,_no_ETOH=19.9±1.01; KD,ETOH=29.8±4.1. (f) Ethanol modulation was assessed for the different variants at a fixed concentration of 25 nM [3H]strychine. α1WT: (in % of Bmax) no ETOH: 44±5, ETOH: 77+4 (**P<0.01, t-test); α1S267N: (in % of Bmax) no ETOH: 61±4, ETOH: 66±3; α1S267Y: (in % of Bmax) no ETOH: 64±7, ETOH: 50±3 (*P<0.05, t-test).
Discussion
In this study, we identified the novel hyperekplexia allele GLRA1(S267N) that affects the ethanol modulatory site within the glycine receptor transmembrane segment 2 of the α1-subunit. This disease-associated GLRA1 allele encodes the substitution of a serine for an asparagine residue that has been characterized in previous in vitro studies to affect the GlyR modulation by alcohols and volatile anesthetics.8 Moreover, transfer of the mutation α1S267Q into knock-in mice produced all hallmark features of dominant hyperekplexia. This indicates that not only ethanol modulation is linked to position α1S267, but also motor function is influenced in the animal model.17
A number of mutations associated with dominant hyperekplexia have been described within TM2.5 According to current structural models, α1S267, the neighboring α1Q266 and α1V260M are localized to the TM2 domain of the GlyR.18 Impairments found in these receptor mutants are similar: notably, the reduced sensitivity to glycine as well as the conversion of β-alanine and taurine into competitive antagonists.13 It should be noted that the conversion of the endogenous GlyR agonists, β-alanine and taurine, to functional antagonists carries the potential to further aggravate the dysfunction associated with hyperekplexia.
A transgenic mouse carrying another ethanol resistant substitution, α1S267Q, exhibits diminished sensitivity to ethanol in behavioral tests,19 indicating that some effects of ethanol can be correlated to a defined site in the glycine receptor. Heterozygous animals (α1/α1S267Q) display enhanced acoustic startle reactions, thus reflecting the dominant effect of the mutation. Upon heterologous expression, α1S267 substitutions differ with respect to their electrophysiology. In α1S267N receptors, we observed an increased EC50 for glycine and no change in Imax, while α1S267Q channels exhibit a normal EC50 and reduced Imax.17 The phenotypic presentation in both cases, however, is compatible with dominant hyperekplexia. While ethanol potentiation was virtually absent in HEK293 expressed homomeric α1(S267N) receptors, some ethanol sensitivity is retained in Xenopus oocyte expression, as potentiation depends on the expression system.14, 17
This study shows that a disease-related receptor allele harbours the potential to modify drug responses in affected patients by reducing GlyR affinity and efficacy. Given the low phenotypic penetrance of the disease allele GLRA1(S267N) in one of our patients, however, this allelic polymorphism may not be clinically apparent. Hence, idiosyncratic drug responses may be attributed to an as yet unrecognized disease allele.
References
Shiang R, Ryan SG, Zhu YZ et al: Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet 1993; 5: 351–358.
Eulenburg V, Becker K, Gomeza J et al: Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun 2006; 348: 400–405.
Rees MI, Lewis TM, Kwok JB et al: Hyperekplexia associated with compound heterozygote mutations in the beta-subunit of the human inhibitory glycine receptor (GLRB). Hum Mol Genet 2002; 11: 853–860.
Rees MI, Harvey K, Pearce BR et al: Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet 2006; 38: 801–806.
Lynch JW : Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 2004; 84: 1051–1095.
Becker K, Hohoff C, Schmitt B et al: Identification of the microdeletion breakpoint in a GLRA1null allele of Turkish hyperekplexia patients. Hum Mutat 2006; 27: 1061–1062.
Lobo IA, Harris RA : Sites of alcohol and volatile anesthetic action on glycine receptors. Int Rev Neurobiol 2005; 65: 53–87.
Mihic SJ, Ye Q, Wick MJ et al: Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 1997; 389: 385–389.
Humeny A, Bonk T, Becker K et al: A novel recessive hyperekplexia allele GLRA1 (S231R): genotyping by MALDI-TOF mass spectrometry and functional characterisation as a determinant of cellular glycine receptor trafficking. Eur J Hum Genet 2002; 10: 188–196.
Breitinger HG, Villmann C, Becker K et al: Opposing effects of molecular volume and charge at the hyperekplexia site alpha 1(P250) govern glycine receptor activation and desensitization. J Biol Chem 2001; 276: 29657–29663.
Tang P, Mandal PK, Xu Y : NMR structures of the second transmembrane domain of the human glycine receptor alpha(1) subunit: model of pore architecture and channel gating. Biophys J 2002; 83: 252–262.
Grudzinska J, Schemm R, Haeger S et al: The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 2005; 45: 727–739.
Rajendra S, Lynch JW, Pierce KD et al: Mutation of an arginine residue in the human glycine receptor transforms beta-alanine and taurine from agonists into competitive antagonists. Neuron 1995; 14: 169–175.
Valenzuela CF, Cardoso RA, Wick MJ et al: Effects of ethanol on recombinant glycine receptors expressed in mammalian cell lines. Alcohol Clin Exp Res 1998; 22: 1132–1136.
Ye Q, Koltchine W, Mihic SJ et al: Enhancement of glycine receptor function by ethanol s inversely correlated with molecular volume at position 267*. J Biol Chem 1998; 273: 3314–3319.
Mascia MP, Mihic SJ, Valenzuela CF et al: A single amino acid determines differences in ethanol actions on strychnine-sensitive glycine receptors. Mol Pharmacol 1996; 50: 402–406.
Findlay GS, Phelan R, Roberts MT et al: Glycine receptor knock-in mice and hyperekplexia-like phenotypes: comparisons with the null mutant. J Neurosci 2003; 23: 8051–8059.
Ma D, Liu Z, Li L et al: Structure and dynamics of the second and third transmembrane domains of human glycine receptor. Biochemistry 2005; 44: 8790–8800.
Findlay GS, Wick MJ, Mascia MP et al: Transgenic expression of a mutant glycine receptor decreases alcohol sensitivity of mice. J Pharmacol Exp Ther 2002; 300: 526–534.
Acknowledgements
We appreciate the contributions of our patients and family members to the study. We thank Renate Fäcke-Kühnhauser, Marina Wenzel and Rosa Weber for expert technical assistance and Dr Carmen Villmann for a critical reading of the manuscript. This work was supported by Deutsche Forschungsgemeinschaft (BE 1138/5-3), the German Israeli Foundation for Research and Scientific Development (GIF 673), and Fonds der Chemischen Industrie (160–43).
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure
The authors have reported no conflicts of interest.
Rights and permissions
About this article
Cite this article
Becker, K., Breitinger, HG., Humeny, A. et al. The novel hyperekplexia allele GLRA1(S267N) affects the ethanol site of the glycine receptor. Eur J Hum Genet 16, 223–228 (2008). https://doi.org/10.1038/sj.ejhg.5201958
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201958
Keywords
This article is cited by
-
A novel nonsense autosomal dominant mutation in the GLRA1 gene causing hyperekplexia
Journal of Neural Transmission (2018)
-
The impact of human hyperekplexia mutations on glycine receptor structure and function
Molecular Brain (2014)
