
Abstract
Increases in the number of allelic malformation syndromes have led to their classification according to their pathogenesis rather than their clinical specific phenotype. TP63 (also known as TP73L) mutations have been identified in several such syndromes characterized by autosomal dominant transmission and various combinations of ectodermal dysplasia, limb malformations and orofacial clefting. TP63 has not yet been implicated in early aging phenotype in humans, even though p63 activates a program of cellular senescence and p63-compromised mice display features of accelerated aging. We report on a family with four affected adult females presenting with Rapp–Hodgkin syndrome (RHS), an autosomal dominant clinical entity that associates anhidrotic ectodermal dysplasia with cleft lip and palate. Features between RHS and EEC syndrome (ectrodactyly, ectodermal dysplasia and cleft lip/palate) have led to the recent identification of mutations in the TP63 gene, located on 3q27, in this condition. Our patients present typical clinical features of RHS, but also ophthalmic anomalies such as corneal dystrophy and premature menopause (around 30 years). The latter findings have never been reported in this condition, and could be secondary to a new TP63 deletion that has been identified in this family.
Similar content being viewed by others

Introduction
The Rapp–Hodgkin syndrome (RHS, MIM 129400) is a rare form of autosomal dominant anhidrotic ectodermal dysplasia.1 The RHS phenotype includes hypohydrosis, cleft lip/palate, small mouth, narrow nose, thin wiry hairs progressing to alopecia in adults, absent or sparse eyebrows and eyelashes, oligodontia or anondotia, hypoplastic nails, abnormal lacrimal ducts, deformed ears and ear canals, and genitourinary anomalies such as hypospadias in males.2 Overlapping features with ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC or Hay–Wells syndrome, MIM 106260) and ectrodactyly, ectodermal dysplasia, cleft lip/palate syndrome (EEC, MIM 129900) have been previously reported.3, 4 Mutations of the TP63 gene have been identified in the following human malformation conditions: EEC syndrome,5, 6 AEC syndrome,7 limb mammary syndrome (MIM 603543),8 acro-dermato-ungual-lacrimal-tooth syndrome (ADULT, MIM 103285),9 nonsyndromic split-hand/foot malformation (MIM 183600),10 isolated cleft lip/palate11 and RHS.12 Most of these entities are characterized by limb abnormalities that fit the split-hand/split-foot spectrum, ectodermal dysplasia affecting the hair, teeth, nails and sweat glands, and other malformations involving the face, the eyes and the urogenital system. Genotype–phenotype correlations have been identified in each of these syndromes.7 Other conditions such as lacrimo-auriculo-dento-digital (LADD) syndrome, ectrodactyly cleft palate syndrome and curly hair ankyloblepharon nail dysplasia syndrome have been suggested to be caused by altered TP63 function but no TP63 mutation in these latter conditions has been identified so far. Since then, mutations in FGF10 and other components of the FGF signalling pathway have been identified in LADD syndrome.13 The TP63 gene is a p53 tumor suppressor gene homolog and encodes TP63 that is a transcription factor. TP63 has two transcription initiation sites and is subject to extensive alternative splicing, giving rise to at least six isotypes having various and opposing activities (Figure 1).12, 14 One class of p63 isoforms contains the N-terminal transactivation domain (TA α, β, γ), homologous to that of p53, whereas the other class of p63 isoforms lacks this domain (ΔN α, β, γ) but is still transcriptionally active.15

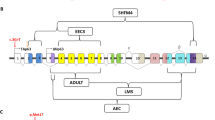
Scheme of P63 gene. (a) Genomic organization of the P63 gene showing the two transcriptional start sites (arrows) and the various splicing. Exon 10′ (previously referred to as exon 15) is located immediately after exon 10.12, 14 The position of the mutation detected in cases III-1 and IV-3 is indicated. (b) Wild-type and mutated α-isoforms. Located in the transactivation inhibitory domain, c.1783delC mutation affects only the α-P63 isoforms.
The balance between these two groups influences proper epidermal morphogenesis and the role of p63 in stem cell maintenance and differentiation seems to be the basis for most of the defects. Indeed, only Tap63 isoforms are expressed in the uncommitted surface ectoderm, whereas ΔNp63 isoforms are expressed after the surface ectoderm has committed to a stratification program.15 The C-terminus of p63 contains a sterile alpha motif (SAM) domain involved in protein–protein interactions (ie binding p63 to the RNA splicing protein, apopbec-1 binding protein), in homo and hetero oligomerization via SAM domains, and a transactivation inhibitory domain (TID), which may have a repressive role in balancing the effects of the different p63 isoforms.16, 17 As the expression of p63 is prominent in the proliferating epithelial basal cell layers of the epidermis in adult humans and mice, the effect of p63 mutations is to disrupt normal ectoderm formation or function, leading to ectodermal anomalies. Embryonic expression of p63 in mice is prominent in various tissues, including corneal epithelial stem cells.18, 19, 20 Homozygous p63−/− mice exhibit craniofacial abnormalities, limb truncations and absence of epidermal appendages.19
Like epidermis, Müllerian vaginal epithelium undergoes columnar to stratified squamous trans-differentiation during development,21 but the Müllerian duct is not regulated by the sequential TA to ΔN expression of p63 isoforms.22 Cervical/vaginal adenosis and reduced fertility in p63+/− mice has already been reported.23 TAp63 seems to be highly expressed in developing primordial germ cells/oocytes, but p63−/− ovaries and oocytes develop normally when grafted in overiectomized adult females. Recently, it has also been shown that TAp63 protects the female germ line during meiotic arrest.24
p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Indeed, p63+/− mice display features of early aging such as lack of skin homeostasis and the inability to maintain hair,23 and defects were identified in their corneal epithelium (H Vogel and AA Mills, personal communication).
We report on a three-generation family with at least four affected individuals, presenting with typical RHS, and new age-related features, which had not been reported earlier in this condition, that is corneal dystrophy and premature menopause. A new TP63 deletion has been identified in this family. We discuss the role of TP63 in this early-aging phenotype and the possible consequences of the identified deletion in our cases.
Materials and methods
Subjects
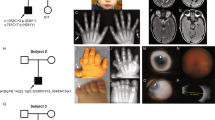
The cases were identified at the genetic clinic of Le Mans Hospital, France. Family histories were taken and blood was collected for DNA analysis, after signed informed consent. RHS was identified in two patients from a large family with at least four affected individuals (Figure 2).

Pedigree of the family.
Case IV-3 was a 42-year-old woman, who presented with the association of sparse, uncombable and straw-like hair, alopecia areata, scanty eyebrows and eyelashes, corneal dystrophy type Groenouw, which began at 40 years, hypodontia, cleft palate and macroglossia, limited sweating, absent tears and mammary hypoplasia (Figure 3a and b). Nails were normal. Puberty started at the age of 13. She did not have any children and presented with premature menopause from the age of 28. Hormonal investigations revealed elevated FSH (72 UI/l) and E2 (32 pg/ml) and progesterone (0.2 ng/ml). At 42 years of age, her vision was altered with 5/10 on each eye. Her elder sister, case IV-2, was also affected and committed suicide. Alopecia started at the age of 17, and hair was uncombable. She also presented with sparse eyebrows and eyelashes, absent tears, corneal dystrophy type Groenouw and Hashimoto thyroditis. Puberty started at 13. Her mother, case III-1, was seen in clinic at the age of 64 years. She also presented with alopecia areata from the age of 20, which became ‘totalis’ at the age of 50 (Figure 3c). Axillary and pubic hair became absent around the age of 20. She also presented with multiple dental infections, and most of her teeth were extracted. Mild deafness appeared recently. She presented with sparse eyebrows and eyelashes, corneal dystrophy type Groenouw, which started at 45 years and absent tears. Nails were dysplastic (Figure 3d). Puberty started at the age of 13, and premature menopause occurred at the age of 35. Breasts were hypoplastic. At the age of 64 years, her vision level was 7/10 on each eye.

(a and b) Facial appearance of case IV-3. Note alopecia aerata and scanty eyebrows. (c) Facial appearance of case III-1. Note alopecia totalis. (d) Hand of case III-1. Note short fourth and fifth metacarpals and dysplastic nails.
II-1 was said to present early alopecia and premature menopause at 28. None of the last two patients presented with either cleft lip or palate, but the clinical findings were consistent with RHS.
Mutational analysis
P63 analysis was conducted on genomic DNA extracted from peripheral blood samples using the EZ1 DNA blood kit with the BioRobot® EZ1 (Qiagen, Courtaboeuf, France). The 16 exons of P63 gene and their flanking intronic regions were amplified by polymerase chain reaction (PCR) and directly sequenced on an ABI 310 genetic analyzer (Applied Biosystems, Courtaboeuf, France) (primer sequences and PCR experimental conditions are available on Supplementary data). The position of mutation is given according to the published TAp63α sequence (GenBank Accession no. AF075430).12
Results
In cases III-1 and IV-3, we detected at codon 595, within exon 14 of the TP63 gene, a heterozygous single nucleotide deletion (c.1783delC, p.Arg595GlyfsX70) (Figure 1, 4). This mutation, which has not previously been described, results in a modification of the reading frame and leads to a stop codon at base 1992 (TAA), 65 bp downstream to the canonic TGA stop. The mutant protein presented the addition of 69 missense amino acids after codon 594 and is 22 amino acids longer than the wild-type protein (Figure 5).

P63 molecular study. Electrophoregram of the proband (a) shows a heterozygous single nucleotide deletion, which is not present in the control (b).

Predicted amino-acid sequence resulting from c.1783delC mutation. c.1783delC leads to the formation of 69 dowstream missense amino acids and a mutant protein that is 22 amino acids longer than the wild-type protein (bolt type indicates the new amino-acid sequence resulting from the frameshift).
Discussion
Here, we report on three patients presenting with typical features of RHS associated with corneal dystrophy and premature menopause, which, to our knowledge, have not been previously reported in this condition. This clinical association could be fortuitous, but this is unlikely, as the eye and endocrinological findings are always segregating with the ectodermal dysplasia in this family.
Corneal dystrophy type Groenouw has been identified in all three cases. Unfortunately, BIGH3 molecular analysis has not been performed and we can not exclude that this eye anomaly could therefore be linked with a mutation of this latter gene, and not with RHS.25 It has been shown that p63 is expressed within the cornea, but its exact function in this tissue remains unclear20 and whether the corneal dystrophy presented by our three cases is linked with the TP63 deletion has to be confirmed.
No other cause of premature menopause was identified in these patients. Premature menopause or primary ovarian failure (POF) can be secondary to a variety of causes including autoimmunity, toxins, drugs, as well as genetic defects. X chromosome abnormalities represent the major cause of primary amenorrhea (ie Turner syndrome or Fragile X syndrome), but CDG syndrome, galactosemia, Blepharophimosis-Ptosis-Epicanthus Inversus syndrome, Pseudohypoparathyroidism, FSH or LH receptor mutations and BMP15 mutations can also be responsible for POF.26, 27, 28 X-inactivation patterns have not been studied in this family, but as it has been previously demonstrated that ΔN p63 is an ectoderm-specific direct transcriptional target of Bmp signalling,29 there might be a molecular connection with POF. Indeed, premature menopause could be an early-ageing feature associated with P63 dysfunction but the molecular mechanisms underlying the consequences of the identified P63 deletion, responsible for a gain of function in our cases, remains to be identified.
It is known that P63 deficiency activates a program of cellular senescence and leads to accelerate aging. It has been demonstrated that p63+/− mice have a significant reduction in lifespan and display features of early aging such as inability to maintain skin and hair.23 Characterization of the functional consequences of P63 gene mutations at a molecular and cellular level is likely to provide further insight into the clinical spectrum, and defining genotype–phenotype correlation is still needed to improve understanding of the various developmental pathways influenced by wild-type or mutant P63. The mutation reported here is located within the C-terminal region of P63. This localization is in accordance with previously identified mutations in RHS.18 As this mutation is here located in the TID, it only affects the two α-isotypes of P63 (TAp63α and ΔNp63α), whereas the β- and γ- isoforms remain intact (Figure 1). A number of frameshift mutations in the TID of P63 have been previously described and result in either RH, limb-mammary or AEC syndrome.10, 18, 30 The reason for this clinical variability is unclear and may be secondary to modifying alleles.31 One of the previously reported mutations in RHS, c.1787delG, results in a mutant protein, which is very similar, except by one amino acid, to our cases.32 However, corneal dystrophy and premature menopause were not associated in this case.
As the TA isoforms are able to activate in vitro p53 target genes and the ΔN isoforms have dominant activities and constitute transcriptional repressors,12 the mutation we describe here could affect the TAp63α isoform and be responsible for a gain of transactivation activity, whereas the dominant-negative effect of ΔNp63α, the major TP63 isotype in epithelial cells, which acts in the maintenance of epithelial stem cells proliferative capacity, could be abolished.4, 12, 33 Yang and McKeon34 have shown that cells that no longer express ΔNp63α lose the capacity of regeneration proliferation and are committed to differentiation. Then, this mutation may also indirectly affect the β- and γ-p63 isoforms by losing the trans-repressive activity of the TID12, 16 and probably deregulates the balance between the different P63 isoforms.18
Although the precise functional consequences of P63 gene pathology need further assessment, the recent awareness of the molecular basis of RHS and other ectodermal dysplasias such as Hay–Wells, ADULT or EEC syndromes with clinical overlap represents useful progresses in understanding and developing a classification of this complex group of autosomal dominant inherited disorders. Determining how the balance of gain or loss of function is affected by specific P63 mutations will be the next step, as well as further studies with mice models that will help towards a better understanding of our findings. Expression studies with our cases' deletion have not been performed, but it would certainly be interesting, as non-previously reported clinical features, such as premature menopause and corneal dystrophy, are associated with the typical RHS phenotype in this family.
Accession codes
References
Rapp RS, Hodgkin WE : Anhidrotic ectodermal dysplasia: autosomal dominant inheritance with palate and lip anomalies. J Med Genet 1968; 5: 269–272.
Moerman P, Fryns JP : Ectodermal dysplasia, Rapp–Hodgkin type in a mother and severe ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC) in her child. Am J Med Genet 1996; 63: 479–481.
Cambiaghi S, Tadini G, Barbareschi M, Menni S, Caputo R : Rapp–Hodgkin syndrome and AEC syndrome: are they the same entity? Br J Dermatol 1994; 130: 97–101.
Celli J, Duijf P, Hamel BC et al: Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999; 99: 143–153.
Brunner HG, Hamel BC, Van Bokhoven H : The p63 gene in EEC and other syndromes. J Med Genet 2002; 39: 377–381.
McGrath JA, Duijf PH, Doetsch V et al: Hay–Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 2001; 10: 221–229.
van Bokhoven H, Hamel BC, Bamshad M et al: p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet 2001; 69: 481–492.
Amiel J, Bougeard G, Francannet C et al: TP63 gene mutation in ADULT syndrome. Eur J Hum Genet 2001; 9: 642–645.
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P : Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 2000; 67: 59–66.
Bougeard G, Hadj-Rabia S, Faivre L, Sarafan-Vasseur N, Frebourg T : The Rapp–Hodgkin syndrome results from mutations of the TP63 gene. Eur J Hum Genet 2003; 11: 700–704.
Leoyklang P, Siriwan P, Shotelersuk V : A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet 2006; 43: e28.
Yang A, Kaghad M, Wang Y et al: p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Rohmann E, Brunner HG, Kayserili H et al: Mutations in different components of FGF signaling in LADD syndrome. Nat Genet 2006; 38: 414–417.
Keyes WM, Vogel H, Koster MI et al: p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc Natl Acad Sci USA 2006; 103: 8435–8440.
Koster MI, Roop DR : Transgenic mouse models provide new insights into the role of p63 in epidermal development. Cell Cycle 2004; 3: 411–413.
Serber Z, Lai HC, Yang A et al: A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol 2002; 22: 8601–8611.
Fomenkov A, Huang YP, Topaloglu O et al: P63 alpha mutations lead to aberrant splicing of keratinocyte growth factor receptor in the Hay–Wells syndrome. J Biol Chem 2003; 278: 23906–23914.
van Bokhoven H, Brunner HG : Splitting p63. Am J Hum Genet 2002; 71: 1–13.
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A : p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999; 398: 708–713.
Pellegrini G, Dellambra E, Golisano O et al: p63 identifies keratinocyte stem cells. Proc Natl Acad Sci USA 2001; 98: 3156–3161.
Kurita T, Cunha GR : Roles of p63 in differentiation of Mullerian duct epithelial cells. Ann N Y Acad Sci 2001; 948: 9–12.
Kurita T, Cunha GR, Robboy SJ, Mills AA, Medina RT : Differential expression of p63 isoforms in female reproductive organs. Mech Dev 2005; 122: 1043–1055.
Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA : p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev 2005; 19: 1986–1999.
Suh EK, Yang A, Kettenbach A et al: p63 protects the female germ line during meiotic arrest. Nature 2006; 444: 624–628.
Munier FL, Korvatska E, Djemai A et al: Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 1997; 15: 247–251.
Goswami D, Conway GS : Premature ovarian failure. Hum Reprod Update 2005; 11: 391–410.
Di Pasquale E, Beck-Peccoz P, Persani L : Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am J Hum Genet 2004; 75: 106–111.
Di Pasquale E, Rossetti R, Marozzi A et al: Identification of new variants of human BMP15 gene in a large cohort of women with premature ovarian failure. J Clin Endocrinol Metab 2006; 91: 1976–1979.
Bakkers J, Hild M, Kramer C, Furutani-Seiki M, Hammerschmidt M : Zebrafish DeltaNp63 is a direct target of Bmp signaling and encodes a transcriptional repressor blocking neural specification in the ventral ectoderm. Dev Cell 2002; 2: 617–627.
Dianzani I, Garelli E, Gustavsson P et al: Rapp–Hodgkin and AEC syndromes due to a new frameshift mutation in the TP63 gene. J Med Genet 2003; 40: e133.
Bertola DR, Kim CA, Albano LM, Scheffer H, Meijer R, van Bokhoven H : Molecular evidence that AEC syndrome and Rapp–Hodgkin syndrome are variable expression of a single genetic disorder. Clin Genet 2004; 66: 79–80.
Chan I, McGrath JA, Kivirikko S : Rapp–Hodgkin syndrome and the tail of p63. Clin Exp Dermatol 2005; 30: 183–186.
Yang A, Schweitzer R, Sun D et al: p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398: 714–718.
Yang A, McKeon F : P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol 2000; 1: 199–207.
Acknowledgements
We are very grateful for the advices that Professor Alea Mills gave us throughout the elaboration of this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Holder-Espinasse, M., Martin-Coignard, D., Escande, F. et al. A new mutation in TP63 is associated with age-related pathology. Eur J Hum Genet 15, 1115–1120 (2007). https://doi.org/10.1038/sj.ejhg.5201888
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201888
Keywords
This article is cited by
-
Heterozygous TP63 pathogenic variants in isolated primary ovarian insufficiency
Journal of Assisted Reproduction and Genetics (2023)
-
Structural diversity of p63 and p73 isoforms
Cell Death & Differentiation (2022)
-
The p63 C-terminus is essential for murine oocyte integrity
Nature Communications (2021)
