
Abstract
The Rubinstein–Taybi syndrome (RTS) is a rare autosomal-dominant disease associated with 10–15% of cases with 16p13.3 microdeletions involving the CREB-binding protein gene (CREBBP). We used array-comparative genomic hybridization and Quantitative multiplex fluorescent-PCR (QMF-PCR) to search for dosage anomalies in the 16p13.3 region and the CREBBP gene. We first constructed a microarray covering 2 Mb that carries seven BAC and 34 cosmid clones, as well as 26 low-molecular-weight probes (1000–1500 bp) that are spread along the CREBBP gene. To increase further the resolution inside the CREBBP gene, we used QMF-PCR assays providing a 7 kb resolution. The deletions characterized in this work extended between as little as 3.3 kb and 6.5 Mb. Some deletions were restricted to just a few exons of CREBBP, some deleted either the 5′ or the 3′ end of the gene plus adjacent genomic segments, others deleted the whole gene away. We also identified a duplication of exon 16. We showed that CREBBP dosage anomalies constitute a common cause of RTS. CREBBP high-resolution gene dosage search is therefore highly recommended for RTS diagnosis. No correlation was found between the type of deletion and the patients’ phenotype. All patients had typical RTS, and there was no particular severity associated with certain alterations.
Similar content being viewed by others


Introduction
The Rubinstein–Taybi syndrome (RTS, [MIM 180849]) is an autosomal-dominant disease that occurs in 1/125 000 births and is characterized by growth retardation and psychomotor development delay, broad and duplicated distal phalanges of thumbs and halluces, a typical facial dysmorphism and an increased tumoral risk. 1 RTS is due to mutations in the CREB-binding protein gene (CREBBP commonly referred to by its shorter acronym CBP).2, 3, 4 Microdeletions including the CREBBP gene are known to account for about 10–15% of RTS cases.2, 5, 6, 7 Our latter study7 clearly showed that CREBBP deletions are heterogeneous in size as well as in position in the gene, since some delete the whole of the CREBBP gene as well as flanking regions away, while others are restricted to intragenic segments.
To perform a comprehensive search for both large and small deletions in patients with the RTS, we have now used two complementary techniques: comparative genomic hybridization on microarrays (array-CGH)8 and quantitative multiplex fluorescent-PCR (QMF-PCR).9 Our data show that gene dosage anomalies of the CREBBP gene span between 3.3 kb and 6.5 Mb, and constitute a more common cause of RTS than was initially thought.
Materials and methods
Rubinstein–Taybi syndrome patients
Patients expressed a typical RTS phenotype, assessed by a detailed clinical questionnaire, photographs, X-rays, and medical records. This study followed ethical guidelines and was authorized by the Comité pour la Protection des Personnes (CPP) of Bordeaux.
Array comparative genomic hybridization
High-molecular-weight clones
A set of seven BACs and 34 cosmids covering 2 Mb in the 16p13.3 region was assembled on the basis of publicly available maps (NCBI build 35 http://www.ncbi.nlm.nih.gov/mapview/ and http://genome.ucsc.edu/cgi-bin/hgGateway) and of our previous work.3 This set of clones compares to two published contigs10, 11 as follows. Our contig extends from AC004493 starting at 2723 Mb from 16pter to AC020663 ending at 4851 Mb from 16pter. The contigs described in Giles et al10 and Bartsch et al11 extend from RT193 (starting at about 3459) to PAC43I6 (ending at about 4575), and from AC93525 (start: 2425) to AC007223 (end: 6616), respectively.
Details about 11q14.3 clones12 and controls from other chromosomal regions (T1, T2, T3, T4, T5, T6, T7) are available from the authors.
Low-molecular-weight clones
Twenty-eight repeat-free targets named aCBPx ranging in size from 1000 to 1500 pb were selected after masking the repeated sequences using the repeat masker software (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker/) (see Supplementary Table 1 for targets’ characteristics). Targets located centromeric to AC020663 are presented in Supplementary Table 2. Seventeen control targets (C1–C17) were selected in various genomic regions (details available from the authors).
Microarray manufacturing and analysis was described by Stef et al13 for low-molecular-weight (LMW) targets; procedures for high-molecular-weight (HMW) targets are available from the authors. Each target was represented at least eight times on the array. Microarrays were scanned with a G2565B scanner (Agilent Technologies). Normalization of values obtained was done using controls T1–T7 for the HMW clones and using controls C1–C17 for the LMW clones.
Quantitative multiplex fluorescent PCR
Twenty-nine PCR assays were designed within a 203 kb interval that includes the CREBBP gene (Supplementary Table 3) and assembled into four sets (Supplementary Table 4). Two control loci were DSCR1 exon 4 (chromosome 21) and coagulation Factor IX (F9) exon 5 (chromosome X). Quantitative multiplex fluorescent PCR was performed as in Niel et al;9 experimental details are available from the authors. Each patient was analysed in triplicate.
Results
Characterization of large deletions in patients with RTS
We have constructed a microarray that covers 2 Mb centred on the CREBBP gene. This microarray harbours 34 cosmids and 7 BAC clones (HMW) and 28 LMW (1–1.5 kb long) repeat-free fragments spread over the CREBBP gene and immediately adjacent sequences (Supplementary Figure 1 and Supplementary Table 1).
Array-comparative genomic hybridization (array-CGH) was used to characterize further large deletions that had previously been observed (patients p4, p9, p31, p34, p59, p73, p197)3, 7 and to identify new ones (patients p218, p219). DNA of RTS patients was hybridized against DNA from a patient (CG) harbouring an 11q14.3 deletion12 as an internal deletion control. Results obtained are shown in Table 1 (HMW targets), and in Supplementary Table 5 (LMW targets). See Supplementary Figure 2 for representative examples of microarray results.
Deletions were very heterogeneous in size and position. We were able to determine the extent of most of them. Deletions removing entirely the CREBBP gene plus flanking segments were observed in patients p219 (about 590 kb) and p59 (about 860 kb).
The farthest centromeric BAC clone (AC020663) was deleted in patients p4 and p218. We therefore designed additional LMW targets at increasing distances from AC020663 towards the centromere (Supplementary materials Table 2). In patient p218, the deletion breakpoint was between LMW 6.42 and LMW 7.45 (Supplementary materials Table 6), that is in an interval located between 2560 and 3550 kb away from exon 1 of the CREBBP gene. The deletion therefore encompassed between 2.4 and 3.9 Mb. In patient p4, the deletion breakpoint laid between LMW 10.1 and 10.2 (Supplementary materials Table 6). This deletion therefore encompassed about 6.5 Mb. These data were confirmed by quantitative multiplex fragment-PCR9 assays derived inside the LMW targets (data not shown).
Identification of small intragenic rearrangements
The microarray clearly showed the existence of deletions limited to one or two cosmids in some patients. This suggested that even smaller rearrangements might be identified. Indeed a deletion of a single target, aCBP10, was found in patient p7 (Supplementary Table 5). We therefore turned to quantitative multiplex PCR, a less expensive approach than array-CGH, to search for small rearrangements. Twenty-nine PCR assays (named qCBP2 to qCBP21; see Supplementary Figure 1e) were designed over a 203 kb genomic segment including the CREBBP gene, allowing us to reach a 7 kb resolution inside the gene. The assays were grouped into four multiplex PCR sets (Supplementary Table 4).
Twenty patients with characteristic features of RTS in whom no point mutation had been identified in spite of a comprehensive investigation were then analysed. Deletions were identified in patients p15, p20, p115, p246, and a duplication in patient p62 (Table 2). These data were confirmed by array-CGH analysis (see Supplementary Figure 3 for patients p15, p20, p62). All of the deletions observed were small, and took between one and three exons away. The duplication in patient p62 contains exon 16. RT-PCR analysis, followed by both restriction and sequence analysis, indicated that exon 16 is duplicated in tandem in the CREBBP RNA (r.3259–3448dup) (data not shown). This duplication introduces a frameshift in the translation, with the appearance of a stop codon at the 13th codon into the second copy of exon 16 (P.Ile1084Asn fsX13). This duplication hence leads to a truncated CREBBP protein that lacks the second half of the protein.
Discussion
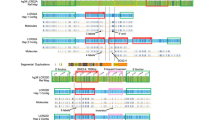
We have used array-CGH and QMF-PCR to find gene dosage anomalies of the CREBBP gene and the surrounding 16p13. 3 region in patients with RTS. Our results show that deletion sizes extend from just a few kilobase pairs (3.3 kb in patient p7; data not shown) to 2400–3900 kb in patient p218 and about 6.5 Mb in patient p4. A small deleterious duplication was identified in one patient. Deletions are very heterogeneous in size and in position with respect to the CREBBP gene. These data are summarized in Figure 1.

Summary of deletion results. (a) Large deletions. A schematic representation of the 2 Mb tiling path contig including 34 cosmid (plain lines) and seven BAC (dotted lines) clones is shown at the top. In the middle is shown a physical map including the CREBBP gene. Positions are given in Megabasepairs from the 16p telomere. The patients’ deletions are represented underneath. Patient numbers are indicated on the right. Not deleted regions are indicated by black lines, deleted regions by white lines. (b) Small intragenic gene dosage anomalies. The upper line depicts the CREBBP gene structure (31 exons). The aCBP line indicates the position of the low-molecular-weight targets used in array-CGH with respect to the CREBBP exons. The qCBP line indicates the position of the QMF-PCR assays with respect to the CREBBP exons. The patients’ deletions are represented underneath. Patient numbers are indicated on the right. Not deleted regions are indicated by black lines, deleted regions by white lines; the duplicated area in patient p62 is represented by a hatched box.
Out of the 21 patients analysed (without a previously identified CREBBP mutation), six were found to have an interstitial gene dosage anomaly (ie 28.6%). Among a total of 83 patients with typical features of RTS analysed in our laboratory, the proportion of patients with a deletion was now estimated to be 20.5% (17/83). The CREBBP rearrangements must therefore be searched for by any kind of cost-effective technique such as QMF-PCR, or others (eg the ligation-based MLPA14 or QMPSF15) before a lengthy search for other mutations of CREBBP or of its homologue P30016 is undertaken.
We did not find a phenotypic difference between patients with small or large deletions, and with deletions extending either towards centromeric or telomeric regions with respect to CREBBP. The only noticeable event is that patient p4 who had the largest deletion (6.5 Mb removing the CREBBP gene plus a large centromeric segment) had a very severe form of the disease with choanal atresia, bilateral dilated ureters, hemivertebrae (T9, T11) and hypoplastic vertebrae (T10, T12), and deceased at 34 days of life.
Particular attention was paid to the phenotype of patients p59 and p218 with a deletion including the DNAseI gene located 62 kb on the telomeric side of CREBBP, since Bartsch et al11 described a particularly severe phenotype with a proneness to repeated infections in patients with a deletion of this gene. Both patients have a rather severe, yet typical, form of RTS (facial dysmorphism, abnormalities of the extremities, mental retardation). Patient p59 is a 39-year-old adult with a small size (132 cm), renal agenesis, radial dislocation, and predisposition to tumours (Hodgkins lymphoma, ovarians cysts, numerous spontaneaous and surgical cheloids, pilomatrixoma). Patient p218 has cardiac (atrial septal defect, patent ductus arteriosus), renal (vesical ureteral reflux) and skeletal (pectus excavatum) malformations. All these features, however, have been described in other RTS patients, including patients with CREBBP point mutations. These patients did not have a particularly severe mental retardation, nor were they particularly prone to infections. It is noteworthy that patient p59 did not develop infections after chemotherapy of her Hodgkins lymphoma. The presentation of these two patients therefore does not confirm the phenotype described by Bartsch et al.11
References
Petrij F, Giles RH, Breuning MH, Hennekam RC : Rubinstein–Taybi Syndrome. In: Scriver CR, Beaudet AL, Valle D, Sly WS, (eds): The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, 8th edn, Chapter 248, 2001, pp 6167–6182.
Petrij F, Dauwerse HG, Blough RI et al: Diagnostic analysis of the Rubinstein–Taybi syndrome: five cosmids should be used for microdeletion detection and low number of protein truncating mutations. J Med Genet 2000; 37: 168–176.
Coupry I, Roudaut C, Stef M et al: Molecular analysis of the CBP gene in 60 patients with Rubinstein–Taybi syndrome. J Med Genet 2002; 39: 415–421.
Bartsch O, Schmidt S, Richter M et al: DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein–Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum Genet 2005; 117: 485–493.
Taine L, Goizet C, Wen ZQ et al: Submicroscopic deletion of chromosome 16p13.3 in patients with Rubinstein–Taybi syndrome. Am J Med Genet 1998; 78: 267–270.
Bartsch O, Wagner A, Hinkel GK et al: FISH studies in 45 patients with Rubinstein–Taybi syndrome: deletions associated with polysplenia, hypoplastic left heart and death in infancy. Eur J Hum Genet 1999; 7: 748–756.
Coupry I, Monnet L, Attia AA, Taine L, Lacombe D, Arveiler B : Analysis of CBP (CREBBP) gene deletions in Rubinstein–Taybi syndrome patients using real-time quantitative PCR. Hum Mut 2004; 23: 278–284.
Solinas-Toldo S, Lampel S, Stilgenbauer S et al: Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 1997; 20: 399–407.
Niel F, Martin J, Dastot-Le Moal F et al: Rapid detection of CFTR gene rearrangements impacts on genetic counselling in cystic fibrosis. J Med Genet 2004; 41: e118.
Giles RH, Petrij F, Dauwerse HG et al: Construction of a 1.2 Mb contig surrounding, and molecular analysis of, the human CREB-binding protein (CBP/CREBBP) gene on chromosome 16p13.3. Genomics 1997; 42: 96–114.
Bartsch O, Rasi S, Delicado A et al: Evidence for a new contiguous gene syndrome, the chromosome 16p13.3 deletion syndrome alias severe Rubinstein–Taybi Syndrome. Hum Genet 2006; 120: 179–186.
Goizet C, Coupry I, Rooryck C et al: Molecular characterization of an 11q14.3 microdeletion associated with leukodystrophy. Eur J Hum Genet 2004; 12: 245–250.
Stef M, Simon D, Burgelin I et al: Testing and improving experimental parameters for the use of low molecular weight targets in array-CGH experiments. Hum Mut 2006; 27: 1143–1150.
Koolen DA, Nillesen WM, Versteeg MHA et al: Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 2004; 41: 892–899.
Casilli F, Di Rocco ZC, Gad S et al: Rapid detection of novel BRCA1 rearrangements in high-risk breast-ovarian cancer families using multiplex PCR of short fluorescent fragments. Hum Mutat 2002; 20: 218–226.
Roelfsema JH, White SJ, Ariyurek Y et al: Genetic heterogeneity in Rubinstein–Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 2005; 76: 572–580.
Acknowledgements
This work was supported by the Conseil Régional d’Aquitaine (no. 20010301132), the Fonds Européen de Développement Régional (FEDER no. 2002-2.2.0.1-1208 and 2004217), the Fondation pour la Recherche Médicale, the European Leukodystrophy Association and the Association Française du Syndrome de Costello. We are grateful to the patients and their family, and to the clinicians who referred the patients. We wish to thank Dr C Bilhou-Nabera (Bordeaux) for her help and advices in FISH experiments, Dr C Rothan and J Petit (Villenave d’Ornon) for their help in microarray spotting, Dr F Niel (Clermont –Ferrand) for advices in the QMF-PCR technique, and T Barnetche (Bordeaux) for help in statistical analysis. Dr N Doggett (Los Alamos) kindly provided the LANL 16p13.3 cosmids. Dr M Breuning (Leiden) kindly provided the RT100 clone.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Stef, M., Simon, D., Mardirossian, B. et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein–Taybi Syndrome patients. Eur J Hum Genet 15, 843–847 (2007). https://doi.org/10.1038/sj.ejhg.5201847
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201847
Keywords
This article is cited by
-
A novel CREBBP mutation and its phenotype in a case of Rubinstein–Taybi syndrome
BMC Medical Genomics (2022)
-
Role of the ADCY9 gene in cardiac abnormalities of the Rubinstein-Taybi syndrome
Orphanet Journal of Rare Diseases (2020)
-
Epigenetic Mistakes in Neurodevelopmental Disorders
Journal of Molecular Neuroscience (2017)
-
Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management
Italian Journal of Pediatrics (2015)
-
Characterization of 14 novel deletions underlying Rubinstein–Taybi syndrome: an update of the CREBBP deletion repertoire
Human Genetics (2015)
