
Abstract
We report on a four-generation family with localized subepidermal telangiectasias following Blaschko's lines (angioma serpiginosum). The vascular streaks are present at birth and progress slowly thereafter. In several family members papillomatosis of the entire oesophagus was found to be part of the condition. Mild nail and hair dystrophy added to the resemblance of Goltz–Gorlin syndrome (focal dermal hypoplasia), suggesting that the present condition could be a mild variant. All affected family members are females, there is no increased miscarriage rate, and X-inactivation in affected females is highly skewed, compatible with X-linked dominant inheritance with very early in utero lethality in males. In the family, 11 informative meioses were available to study the segregation of X-chromosome markers. Significant linkage (LOD score 3.31) was found to a region flanked by markers DXS8026 and DXS106 (44–67 Mb from Xpter) that includes the centromere.
Similar content being viewed by others

Introduction
Angioma serpiginosum (OMIM no. 106050) is a congenital skin disorder characterized by capillary ectasias in the superficial papillary dermis, which show clinically as linearly grouped punctate lesions on a mildly erythematous background. The condition was first described by Jonathan Hutchinson in 18891 and named 5 years later.2 The rash has a serpiginous or gyrate pattern that follows Blaschko's dermal migration lines.3 The rash resembles purpura,4, 5, 6 but there are no signs of inflammation or bleeding.7, 8 The vascular capillary nests in the subepidermal papillae are not simple telangiectasias but represent a capillary naevus with both dilatation and proliferation of thick-walled capillaries having slit-like protrusions of capillary lumen into the endothelial lining.9, 10 Only incomplete blanching by pressure can be obtained.11, 12 The rash is otherwise asymptomatic, and the patients' problems are mainly of cosmetic nature. Treatment by pulsed dye laser can be successful.13, 14 Lesions have been found to be confined to isolated body segments,10 especially the lower limbs,11, 15 but sometimes there is a more general distribution. Extensive affection of the skin is rare.4, 12, 16 The palms and soles are not affected, nor is the oral mucosa.
Around 90% of patients are female.15 Familial occurrence is rare: there is no male-to-male transmission reported but two father-to-daughter transmissions are known.11, 12 In the only large family with angioma serpiginosum so far described, four out of six siblings were affected (two brothers and two sisters), and three of them had an affected daughter, suggesting autosomal dominant inheritance.11
We have investigated a Norwegian family with suspected X-linked dominant angioma serpiginosum, the second family ever described with this usually sporadic condition. We have studied the inheritance pattern, the X-inactivation status of healthy and affected females, and carried out linkage analysis and found the location of the gene. A serendipitous finding was that angioma serpiginosum is associated with oesophageal papillomatosis, suggesting that this condition may be a mild variant of the X-linked dominant Goltz–Gorlin syndrome.
Materials and methods
Clinical description
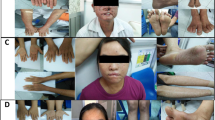
In a four-generation Norwegian family with angioma serpiginosum, all six affected individuals are female, and the rash follows the lines of Blaschko, suggesting X-linked dominant inheritance (Figure 1). The proband (Figure 1: arrow) is a 46-years-old woman with an asymptomatic punctate and linear erythematous rash that had progressed slowly since birth. The rash is localized on the left shoulder, the inside of both arms, on the back, and on the outside and back of the thighs and calves (Figure 2b). The rash has improved after laser treatment. Skin biopsy showed nests of slightly dilated and somewhat thick-walled capillaries in the dermal papillae and upper part of the dermis with areas of hyperkeratosis and no signs of inflammation. The skin is otherwise dry, lips, mouth mucosa, tongue and teeth are normal, and nails are slightly dysplastic with ridges and incisions. The hair is thin and straight. The proband has an 11-years-old daughter with a similar slowly progressive rash, as well as slightly dystrophic nails and a dry skin. In her, the rash is presently found on both arms and the left thigh and leg (Figure 2a, c and d). Only incomplete blanching by pressure can be obtained. The capillary dilatations are partly punctate, partly linear and short (Figure 2). Long telangiectatic vessels have not been seen.

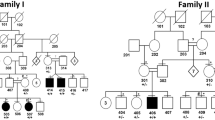
Pedigree, X-inactivation and linkage analysis: there are six affected women in four generations, of whom four are still alive. The result of the X-inactivation analysis is shown above the pedigree symbols: highly skewed X-inactivation was only found in affected women, in one case the analysis was non-informative (N.I.). The markers used and the haplotypes found in the critical region are shown below. The allele sizes are given as observed fragment size (in bp) with the exception of AR, where the size is shown as the number of repeats. All affected family members shared a 23 Mb stretch of X-chromosome DNA that was not found in any unaffected family member, marked by a box. The grey scale refers to the haplotypes found in II–1: The disease-associated haplotype is dark grey, the other light grey. The other haplotypes are contributed by male spouses of affected women; for clarity their genotypes are not shown.

The rash on the back of the left leg of the proband (b) and on the left leg (a, c) and left upper arm (d) of her daughter at age 11 years: a slowly progressive punctate erythema following Blaschko's lines.
A younger sister of the proband also has similar rash, skin and nail findings. She was investigated because of mild retrosternal pain and dysphagia at the age of 30 years, and small papillomatous tumours were found in the pharynx. Endoscopy showed multiple lobulated small tumours of a colour similar to oesophageal mucosa immediately beyond the pharyngoesophageal junction. In the upper half of the oesophagus, the papillomas were confluent, leaving only a small irregular lumen (Figure 3). The number of papillomas decreased towards the distal oesophagus. Computerized tomography showed irregular lumen and increased thickness of the oesophageal wall. Oesophageal manometry, 24 h ambulatory pH monitoring and bronchoscopy gave normal results. Histologically, the papillomas had a central core of vascular connective tissue covered by a squamous epithelium with disturbed maturation (marked variation in size and shape of the nuclei in the superficial layers). Koilocytes (cells with hyperchromatic nuclei and perinuclear halos) were also seen, highly reminiscent of epithelium with human papilloma virus (HPV) infection.17 However, no evidence for HPV infection was found, even by using molecular techniques (in situ hybridization and PCR). The oesophageal findings in this patient have been published, but the authors were at that time unaware that the patient also had angioma serpiginosum.17

Papillomatosis in the upper half of the oesophagus. Histologically, most papillomas had a diameter of 1–4 mm.
Esophagoscopy in the proband showed oesophageal papillomatosis of the same type and distribution as in her sister. The tumours were completely asymptomatic. The mother of the two sisters also has angioma serpiginosum with sparse hair, dry skin and nail changes similar to the proband. Her oesophagus has not been investigated. A sister of mother did have an even more extensive rash. This sister died at age 61 years from coronary heart disease. The proband's grandmother died at age 94 years, and anamnestically she was affected as well. Probably, she was the first in the family with the condition (Figure 1). There is no history of eye problems, infertility or increased spontaneous miscarriage rate in affected females.
Methods
After obtaining informed consent, blood samples were obtained from four affected and seven unaffected family members (Figure 1). X-inactivation status was studied by analysing the methylation pattern of the polymorphic trinucleotide repeat in the first exon of the androgen receptor (AR) gene.18 After digestion with the methylation-sensitive restriction enzyme HpaII, PCR products will be obtained from the inactive X chromosome only. These were separated by capillary electrophoreses on an ABI310 automated sequencer, and analysed by GeneScan software (Applied Biosystems, Foster City, CA, USA).
Linkage analysis was initially carried out by using the 10 cM X-chromosome STR-panel from the ABI PRISM® Linkage Mapping Set (Applied Biosystems). Additional STRs were added in regions with non-informative markers. Using the PC-version of the Rockefeller University linkage programs (www.genemapping.cn/linkhelp.htm), positive LOD scores were found for markers around the X centromere. To confirm linkage and determine the haplotype segregating with disease, fine STR mapping of the pericentromeric region was carried out (Figure 1).
Results
It turned out that affected females had an extremely skewed X-inactivation pattern in blood leucocytes: two showed 100:0, one had 96:4, and one was non-informative due to allele overlap (Figure 1). It was consistently the X-chromosome from the affected mother that was inactivated in affected females, strongly supporting our assumption of X-linked dominant inheritance. Furthermore, X-chromosome linkage analysis showed that a region flanked by DXS8026, 44.0 Mb from Xpter, and DXS106, 67.7 Mb from Xpter, was the only region shared by all affected females and not found in any unaffected males or females (Figure 1). The region included the X centromere. There are 12 informative meioses in the family, of which 11 were available for direct study. After determination of haplotypes (phase), there are 11 meioses in which there is an a priori 0.5 chance for separation of the disease and the haplotype under the null-hypothesis (no linkage). Accordingly, the likelihood that our observation is a chance finding, assuming that the haplotype is inherited from the proband's maternal grandmother, is (½)11=1:2048. The log10 of the odds ratio (2048:1) is 3.31. This is identical to the two point (haplotype and disease) LOD score obtained by the MLINK program at θ=0 (no recombinations). According to the NCBI gene map (build 36.2), there are 275 known and putative genes in this interval. As vascular malformations are the major symptoms of the present condition, we sequenced three genes that theoretically are of importance for angiogenesis: MSN (moesin, important for cell–cell recognition, signalling and cell movement); TIMP1 (matrix metalloproteinase inhibitor) and FKSG43 (intronless gene with some KRIT1 homology). No mutations were found.
Discussion
The present results indicate angioma serpiginosum to be an X-linked dominant entity with male lethality. Papillomatosis of the oesophagus can be part of the syndrome. The HPV-infection-like dysplasia of the squamous epithelium of the oesophagus,17 indicates that the gene might function in the regulation of cell differentiation or apoptosis. We have no indications that the patients have increased risk for oesophageal cancer. In the only other large family with angioma serpiginosum reported so far, the pattern of inheritance is best compatible with autosomal dominant inheritance but does not exclude X-linked dominant inheritance.11 As most patients from the literature are female, and the vascular streaks often follow Blaschko's lines,3, 15 an X-linked dominant acting gene is likely to be the major cause in most sporadic cases. The few described males often have segmental affection,10 suggesting somatic mutations. The lack of extended families indicates that mutations are often incompatible with foetal survival, a notion that is supported by the skewed X-inactivation pattern that we found in leucocytes of the present patients. It is conceivable that our family has an unusual and mild mutation in a gene critical for embryonic survival. The mutation should be sufficiently mild to be compatible with early embryonic survival until the time of X-inactivation, which selects against cells with the mutated X as the active X chromosome. The skewed X-inactivation in leucocytes, the mild ectodermal symptoms and the oesophageal findings in the present patients show that the gene has a more widespread role beyond angiogenesis in the dermal papillae.
There is considerable overlap of the present entity with Goltz–Gorlin syndrome. Goltz–Gorlin syndrome is a sporadically occurring X-linked dominant condition seen only in females. The main features are congenital skin hypoplasia or atrophy following the lines of Blaschko, dysplastic nails, sparse or brittle hair, skin telangiectasias, limb anomalies, and multiple papillomas of the skin and mucous membranes.19 There are also reports of patients with oesophageal papillomatosis,20, 21 or papillomas in the hypopharynx.22 This resemblance leaves open the possibility that angioma serpiginosum with oesophageal papillomatosis is a mild variant of Goltz–Gorlin syndrome that does not affect female fertility to the same degree as in classical Goltz–Gorlin syndrome. Further studies in both angioma serpiginosum and Goltz–Gorlin syndrome are needed to proof or disproof this.
References
Hutchinson J : A peculiar form of serpiginous and infective naevoid disease. Arch Surg 1889; 1: Plate IX.
Crocker HR : Lymphodermia perniciosa. Br J Dermatol 1894; 6: 367.
Gerbig AW, Zala L, Hunziker T : Angioma serpiginosum, a skin change along Blaschko lines? Hautarzt 1995; 46: 847–849.
Barker LP, Sachs PM : Angioma serpiginosum, a comparative study. Arch Dermatol 1965; 92: 613–620.
Ohnishi T, Nagayama T, Morita T et al: Angioma serpiginosum: a report of 2 cases identified using epiluminescence microscopy. Arch Dermatol 1999; 135: 1366–1368.
Cox NH, Paterson WD : Angioma serpiginosum: a simulator of purpura. Postgrad Med J 1991; 67: 1065–1066.
Svendsen E, Olsen P : Angioma serpiginosum. A diagnostic problem. J Norw Med Assoc 1985; 105: 1308–1309.
Erbagci Z, Erbagci I, Erkilic S, Bekir N : Angioma serpiginosum with retinal involvement in a male: a possible aetiological role of continuous cold exposure. J Eur Acad Dermatol Venereol 2004; 18: 238–239.
Kumakiri M, Katoh N, Miura Y : Angioma serpiginosum. J Cutan Pathol 1980; 7: 410–421.
Al Hawsawi K, Al Aboud K, Al Aboud D, Al Githami A : Linear angioma serpiginosum. Pediatr Dermatol 2003; 20: 167–168.
Marriott PJ, Munro DD, Ryan T : Angioma serpiginosum – familial incidence. Br J Dermatol 1975; 93: 701–706.
Sandhu K, Gupta S : Angioma serpiginosum: report of two unusual cases. J Eur Acad Dermatol Venereol 2005; 19: 127–128.
Long CC, Lanigan SW : Treatment of angioma serpiginosum using a pulsed tunable dye laser. Br J Dermatol 1997; 136: 631–632.
Ilknur T, Fetil E, Akarsu S, Altiner DD, Ulukus C, Gunes AT : Angioma serpiginosum: dermoscopy for diagnosis, pulsed dye laser for treatment. J Dermatol 2006; 33: 252–255.
Frain-Bell W : Angioma serpiginosum. Br J Dermatol 1957; 69: 251–268.
Katta R, Wagner A : Angioma serpiginosum with extensive cutaneous involvement. J Am Acad Dermatol 2000; 42: 384–385.
Sandvik AK, Aase S, Kveberg KH, Dalen A, Folvik M, Naess O : Papillomatosis of the esophagus. J Clin Gastroenterol 1996; 22: 35–37.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW : Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Human Genet 1992; 51: 1229–1239.
Gorlin RJ, Cohen MM, Hennekam RCM : Syndromes of the head and neck. New York: Oxford University Press, 2001.
Zala L, Ettlin C, Krebs A : Focal dermal hypoplasia with keratoconus, papillomatosis of esophagus and hidrocystomas (author's transl)]. Dermatologica 1975; 150: 176–185.
Brinson RR, Schuman BM, Mills LR, Thigpen S, Freedman S : Multiple squamous papillomas of the esophagus associated with Goltz syndrome. Am J Gastroenterol 1987; 82: 1177–1179.
Gordjani N, Herdeg S, Ross UH, Grimme H, Kleinschmidt M, Brandis M : Focal dermal hypoplasia (Goltz–Gorlin syndrome) associated with obstructive papillomatosis of the larynx and hypopharynx. Eur J Dermatol 1999; 9: 618–620.
Acknowledgements
We thank Jack Johansen at Førde Hospital for esophagoscopy in the proband, the Diagnostic DNA laboratory at Haukeland University Hospital for linkage analysis, the Department of Pathology at Haukeland University Hospital for histological examination of the skin biopsies, and Helge Boman for helpful discussion of the paper. This study could not have been done without the generous collaboration of the present family.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the memory of Robert J Gorlin (1923–2006)
Rights and permissions
About this article
Cite this article
Blinkenberg, E., Brendehaug, A., Sandvik, A. et al. Angioma serpiginosum with oesophageal papillomatosis is an X-linked dominant condition that maps to Xp11.3–Xq12. Eur J Hum Genet 15, 543–547 (2007). https://doi.org/10.1038/sj.ejhg.5201800
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201800
Keywords
This article is cited by
-
Esophageal papillomatosis: an exceedingly rare disease
Orphanet Journal of Rare Diseases (2023)
-
Goltz–Gorlin (focal dermal hypoplasia) and the microphthalmia with linear skin defects (MLS) syndrome: no evidence of genetic overlap
European Journal of Human Genetics (2009)
-
Reply to Happle
European Journal of Human Genetics (2009)
-
Angioma serpiginosum is not caused by PORCN mutations
European Journal of Human Genetics (2009)
-
An Xp11.23 deletion containing PORCN may also cause angioma serpiginosum, a cosmetic skin disease associated with extreme skewing of X-inactivation
European Journal of Human Genetics (2008)
