
Abstract
Unbalanced translocations, that involve the proximal chromosome 15 long arm and the telomeric region of a partner chromosome, result in a karyotype of 45 chromosomes with monosomy of the proximal 15q imprinted region. Here, we present our analysis of eight such unbalanced translocations that, depending on the parental origin of the rearranged chromosome, were associated with either Prader–Willi or Angelman syndrome. First, using FISH with specific BAC clones, we characterized the chromosome 15 breakpoint of each translocation and demonstrate that four of them are clustered in a small 460 kb interval located in the proximal 15q14 band. Second, analyzing the sequence of this region, we demonstrate the proximity of a low-copy repeat 15 (LCR15)-duplicon element that is known to facilitate recombination events at meiosis and to promote rearrangements. The presence, in this region, of both a cluster of translocation breakpoints and a LCR15-duplicon element defines a new breakpoint cluster (BP6), which, to our knowledge, is the most distal breakpoint cluster described in proximal 15q. Third, we demonstrate that the breakpoints for other rearrangements including large inv dup (15) chromosomes do not map to BP6, suggesting that it is specific to translocations. Finally, the translocation breakpoints located within BP6 result in very large proximal 15q deletions providing new informative genotype–phenotype correlations.
Similar content being viewed by others


Introduction
The proximal long arm of human chromosome 15 (chr 15) is involved in a wide range of structural rearrangements that lead to segmental aneusomy of the 15q11–q13 region. As this region is known to be imprinted, the associated phenotype depends on the parental origin of the rearranged chromosome.
Interstitial deletions of the proximal 15q region, spanning approximately 4 Mb, result in Prader–Willi syndrome (PWS, MIM # 176270) or Angelman syndrome (AS, MIM # 105830), when the deletion occurs on the paternal or maternal chromosome, respectively.1, 2
Interstitial duplications of this region are associated with varying degrees of mental retardation and autistic behaviour, when maternally derived. In contrast, paternally derived duplications are generally associated with a normal phenotype.3, 4, 5, 6
Inverted duplications, known as supernumerary inv dup (15) chromosomes, are common rearrangements of the 15q11–q13 region. Large inv dup (15) chromosomes, which are exclusively of maternal origin, have been reported in patients with facial dysmorphy, mental retardation, severe epilepsy and, in many cases, autistic behaviour.7, 8, 9, 10
Rare intrachromosomal triplications of either maternal or paternal origin also occur in the proximal 15q region and are generally associated with a phenotype analogous to that for large maternally derived inv dup (15).11, 12, 13
Finally, unbalanced translocations have been described with one breakpoint in proximal chromosome 15q and another in the telomeric region of the partner chromosome that in exceptional cases is situated within the TTAGGG telomeric repeats (14, 15, 16, 17 and references therein). Such rare translocations are associated with a karyotype of 45 chromosomes and monosomy of the proximal 15q leading to a PWS or AS phenotype.
With the exception of the unbalanced translocations, all these rearrangements have been shown to occur in preferential regions of chr 15, known as BPs. Five BPs (BP1–BP5) have been characterized in the chromosome 15q11–q13 region.18 The genetic instability in this region has been attributed to relatively large genomic duplications (duplicons) that map in the vicinity of the BPs. Such duplicons are believed to mediate misalignment during meiosis, leading to unequal recombination events. BPs BP1, BP2 and BP3, which are mainly associated with interstitial deletions and duplications, are characterized by duplicons that contain repeats derived from the ancestral HEct domain and RCc1 domain protein 2 gene, as well as a number of other low-copy repeat (LCR15) elements.18, 19, 20 BP4 and BP5, which are most frequently involved in triplications and inv dup (15) chromosomes, are characterized by duplicons that contain only LCR15 elements.18
Nevertheless, no specific BP that characterizes unbalanced translocations of the proximal chromosome 15q onto the telomeric band of a partner chromosome has been described. In this study, we characterized the chr 15 breakpoint from eight unbalanced translocations of this type and demonstrated that four of them are clustered in a reduced 460 kb interval located in the proximal 15q14 band. We also demonstrated the presence of an LCR15-duplicon element at the proximal boundary of that interval. The presence of a cluster of translocation breakpoints at the vicinity of a LCR15-duplicon allowed us to define a new BP which we name (BP6). We also demonstrated that BP6 contains no breakpoint of large inv dup (15) chromosomes, suggesting it is specific to unbalanced translocations. Finally, as the translocation breakpoints that occur in this interval result in a proximal 15q deletion which is larger than usual, we searched for additional clinical signs that correlate to the size of the deletion.
Materials and methods
Patients
We studied eight patients with a karyotype of 45 chromosomes resulting from an unbalanced translocation of the proximal chromosome 15q onto the telomeric band of the partner chromosome. All these patients, therefore, were monosomic for the imprinted proximal 15q region and presented either an AS or a PWS, depending on the parental origin of the deletion.
Three patients with PWS phenotype had been reported previously: Case 1 with a 45,XY, −3, −15, +der(3)t(3;15) (q29;q14) karyotype presented additional features which are uncommon in PWS, including preauricular tags, bilateral clubfoot, persistent foramen ovale and small patent ductus arteriosus, and central visual impairment.21 Case 2, a four-year-old girl with karyotype: 45,XX, −11, −15, +der(11)t(11;15)(q25;q13), presented a more severe developmental delay than usually observed in PWS.22 Case 3 with a 45,XX, −15, −21, +der(21)t(15;21)(q13;q22.3) formula presented signs of cerebral atrophy, malformed ears, and severe mental and psychomotor retardation, and unusual in PWS.23
Four patients were referred to one of us for suspicion of either PWS (case 4 and 6) or AS syndromes (cases 5 and 7): Patient 4 is a female patient who had presented with severe neonatal hypotonia with poor sucking. PWS was diagnosed by both cytogenetic and molecular analyses that revealed a de novo unbalanced translocation: 45, XX, −10, −15, +der (10)t(10;15) (q26;q14), involving the paternal chromosome 15. Four months after birth, the growing delay persisted and microcephaly (−2DS) was noted. At more than 4 years, the patient did not walk alone, had no language and appeared to be severely retarded. Patient 6 is an 11-year-old female with PWS diagnosed at birth because of severe generalized hypotonia, feeding difficulties, and cranio-facial dysmorphy. Hyperphagia developed in early childhood with insatiable appetite and obesity. She was mildly mentally retarded. Cytogenetic and molecular evaluation revealed a de novo unbalanced translocation: 45, XX, −13,−15, +der(13)t(13;15)(p11;q13) originated in the paternal meiosis. Patient 5 is a male patient with AS diagnosed when he was 2 years old, because of global developmental delay, facial dysmorphy, and seizures. The karyotype revealed a de novo unbalanced translocation: 45, XY, −1, −15, +der(1)t(1;15) (q44;q13) and methylation studies demonstrated maternal origin of the translocation. Patient 7 is a 30-year-old male patient diagnosed as AS. The proband presented severe developmental delay (he has never walked alone) and severe mental retardation. He developed generalized seizures at 3 years. His severe phenotype was attributed to complications of neonatal episodes of anoxy. His karyotype is: 45, XY,−15,−22, +der(22)t(15;22) (q13;p11).
Finally, Patient 8 is a 2 weeks- black male, presenting an unbalanced translocation 45,XY, −13, −15, +der(13) t(13;15)(q34;q15) of maternal origin and a not well defined phenotype. The pregnancy was complicated by maternal substance abuse and he presented sloping forehead, small anterior fontanel; overriding sagittal, coronal and lambdoid sutures; cup-shaped ear deforms; very prominent nasal root; abnormal palmar creases; contractures; and increased tone. A fibroblast cell line from this patient (case 8) was obtained from the Coriell Cell Repositories (GM 10329, Camden, NJ, USA).
We also analyzed the breakpoints from 18 cases of supernumerary inv dup (15) chromosomes, in order to compare them with the breakpoints of the unbalanced translocations.
FISH analysis, molecular DNA probes, sequence analysis
Metaphase spreads were obtained with standard protocols, either from short-term blood lymphocyte cultures, or from EBV-transformed lymphoblastoid cell cultures, or again from fibroblast cell cultures. FISH on metaphase chromosomes were used to characterize the breakpoints of both chromosomes involved in each unbalanced translocation.
Different commercial probes were used to determine the breakpoint onto the telomeric region of the recipient chromosomes, as recommended by the manufacturers. A synthetic peptide nucleic acid PNA-(CCCTAA)3 oligonucleotide probe (Dako SA, Trappes, France) was first used to detect consensus telomeric TTAGGG repeats. Several subtelomeric probes were secondarily used based on results obtained from the telomeric repeats probe: the 1q subtelomere 1QTEL10 probe (Abbott Laboratories, Rungis, France); the 11q subtelomere LPT11Q and the 13q subtelomere LPT13Q probes (Cytocell technologies, Cambridge, UK); the 21q subtelomere PTEL21q (Q-Biogene, Illkirch, France).
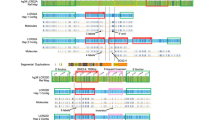
Bacterial artificial chromosomes (BACs) used to narrow the chr 15 breakpoint regions were provided by the Sanger Institute (www.sanger.ac.uk). They were labelled with either biotin-14dCTP (Bioprime DNA labelling system, Life Technologies) or digoxigenin-11dUTP (Highprime, Roche Diagnostics, Meylan, France). This led us to use about 35 BACs clones from chromosome 15 (RPCI-11 and RPCI-13 libraries) and only the most informative ones are summarized in Figure 1. In the different FISH experiments, normal and translocated chr 15 s were identified with either the BAC 327J17 (located at 15q26.2), or the BAC 209K10 (located at 15q21.3).

Schematic representation of the 15q11–15q14 region. The BPs, specific to recurrent rearrangements involving the proximal 15q, are represented on the left of the enlarged chr 15q11–q14 region. On the right are represented: the BACs/markers that allowed us to define the rearrangement breakpoints, the distribution of the eight translocation breakpoints (transloc Case N°) and the distribution of the 18 inv dup (15) breakpoints (Inv dup Case N°). Patients with inv dup (15) chromosomes were distributed as follows: Ten with karyotype: 47, XX, +inv dup (15), cases 1, 2, 3, 5, 6, 7, 9, 11, 13, 17; Eight with karyotype: 47, XY, +inv dup (15), cases 4, 8, 10, 12, 14, 15, 16, 18.
Finally, sequence analysis of the 460 kb interval was performed using the following database and bioinformatic tools:
Entrez Nucleotides database: (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide)
GCG programs (Accelrys)/SeqWeb interface: (http://www.infobiogen.fr)
Human Genome Segmental Duplication Database Assembly; May 2004: http://projects.tcag.ca/cgi-bin/duplication/dupbrowse/human_b35)
Repeat masker Web Server: (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker)
Results
Determination of the translocation breakpoints by standard and molecular cytogenetics
All eight patients had a karyotype with 45 chromosomes, and carried an unbalanced translocation. No chromosomal material other than the proximal 15q region appeared to be deleted in cells from all patients analyzed by high-resolution chromosomal banding.
We first characterized the translocation breakpoint on the partner chromosomes, and the results are reported in Table 1. FISH experiments using the PNA-(CCCTAA)3 probe revealed the presence of interstitial TTAGGG telomere repeats at the junction of the translocated chromosomes in three cases (cases 1, 4, and 6) (Figures 2a and b). This suggested that the breakpoint of these translocations is within the telomeric repeats of the partner chromosome. In the other cases, no interstitial telomere signal was detected on the translocated chromosome, suggesting that the breakpoint on the partner chromosome is proximal to the telomeric repeats. Consequently, FISH was performed with subtelomeric specific probes in order to refine breakpoints on the translocation partner chromosomes. For case 7, the telomeric breakpoint is on the short arm of an acrocentric chromosome containing only repetitive sequences the deletion of which has no phenotypic effect. For the other translocations, we found that the specific 13q subtelomeric probe was present in case 8, but the 11q, 21q, and 1q subtelomeric probes were deleted in cases 2, 3, and 5 respectively. These results demonstrate that, for the majority of the translocations (cases 1, 4, 6, 7, and 8), the phenotype results from the segmental aneusomy of chr 15 alone, as no coding sequence is deleted from the partner chromosome. For the three remaining cases, despite the absence of the subtelomeric specific probe from the recipient chromosome, high-resolution studies show that the deletion is cryptic.

Detection of interstitial telomeric TTAGGG repeats on metaphasic chromosomes of unbalanced translocations. FISH with a Cy3-labelled synthetic PNA-(CCCTAA)3 probe reveals interstitial telomeric repeats, (a) in case 1, on the translocation t(3;15) (red signal, white arrows); (b) in case 4, on the translocation t(10;15) (red signal, white arrow). In this case, the normal and translocated chromosome 15 s are simultaneously identified with BAC 209K10 (green signal), that maps to 15q21.3. Metaphasic chromosomes are counterstained in blue with DAPI.
In order to localize the translocation breakpoints on the chr 15, we hybridized chr 15 specific BAC probes onto metaphases from each patient and the results are summarized in Figure 1. Four translocation breakpoints were scattered between the 15q11.2 and 15q13.1 bands: case 6 (between BACs 446P9 and 1084I9), case 7 (between BACs 142M24 and 10K20), case 5 (within the BAC 665A22), and case 2 (between BACs 665A22 and 483E23). Four other breakpoints (cases 1, 3, 4, and 8) were clustered in a small interval delineated by BACs 64O3 and 150L8, and located within the proximal 15q14 band.
Refined localization of the translocation breakpoints within the 15q14 interval
In order to refine the localization of the four breakpoints clustered within the 15q14 interval, we used BAC clones covering the entire interval and ordered as follows, from proximal to distal: BACs 64O3, 814P5, 83J16, 323I15, and 150L8.
For patient 1, FISH analysis of metaphases using BAC 323I15 revealed a signal of very low intensity on the translocated chr 15 compared to the signal observed on the normal 15 (Figure 3a). This demonstrated that the chr 15 breakpoint was located within the BAC 323I15, and probably at its distal end (Figure 4).

Characterization of the chromosome 15 translocation breakpoints. (a) In case 1, the BAC 323I15 (green signal) shows a very low-intensity signal on the t(3;15) chromosome, compared to the one observed on the normal chromosome 15. This is in agreement with the breakpoint being located within the BAC 323I15. (b) In case 4, the BAC 83J16 (green signal) shows only one FISH signal, located on the normal chromosome 15. This BAC is absent on the t(10;15) chromosome. In both cases, the normal and the translocated chromosome 15 s are identified with BAC 327J17 (red signal), that maps to 15q26.2. Metaphasic chromosomes are counterstained in blue with DAPI.

Schematic representation of the BP6 breakpoint cluster, located within the proximal 15q14 band. For each of the cases 1, 3, 4 and 8, the localization of the chromosome 15 breakpoint relative to the BACs is represented by a vertical dotted line. The large black box adjacent to BAC 64O3 represents the LCR15-duplicon element.
For patient 3, FISH analysis revealed BAC 64O3 to be deleted and BAC 814P5 to be present on the translocated chr 15. Consistent with the fact that BAC 814P5 showed similar sized FISH signals on both the normal and translocated chr 15 s, we mapped the chr 15 breakpoint to the very proximal end of BAC 814P5 (Figure 4).
For patient 4, no FISH signal was detected on the translocated chr 15 using BAC 83 J16, although a normal FISH signal was detected using the 323 I15 BAC (Figure 3b). This suggests that the chr 15 breakpoint is located between the distal boundary of BAC 83J16 and the proximal boundary of BAC 323I15 (Figure 4).
For patient 8, BAC 323I15 revealed no signal on the translocated chr 15, although BAC 150L8 gave an apparently normal-sized FISH signal. This was in agreement with a chr 15 breakpoint being located in the overlapping region of these BACs (Figure 4).
We therefore demonstrated that four translocation breakpoints are located within a reduced interval of 460 kb, which is delineated by the distal boundary of BAC 64O3 and the proximal boundary of BAC 150L8. Although these breakpoints are not identical, their clustering in a small interval suggested that a common mechanism could lead to the occurrence of these rare translocations involving proximal 15q and the telomeric region of the recipient chromosome.
Comparison of breakpoints between the translocations and large inv dup (15)
In order to evaluate whether the small 460 kb interval also contains breakpoints from other chr 15 rearrangements, we determined the breakpoints of 18 large inv dup (15) chromosomes, using FISH with BAC clones delineating BP3, BP4, and BP5 (Figure 1). Indeed, breakpoints from such rearrangements have been shown to occur in all the BPs described on proximal 15q and particularly in the more distal BP4 and BP5. Thus, such large inv dup (15) chromosomes could be expected to have a breakpoint even more distal than BP5, that is, within or around our 460 kb interval. Strikingly, none of the 18 cases studied was found to contain the BAC 489D6 (marker D15S144) that marks the most distal boundary of BP5.18 Indeed, our FISH studies demonstrated that five inv dup (15) had a breakpoint located within BP3 (Figure 1), three had a breakpoint located within BP4 and 10 had a breakpoint within BP5.
These results, which demonstrate that no breakpoints from large inv dup (15) chromosomes are located in the small 460 kb interval, suggest that this interval is specific to translocations.
Searching for DNA sequences favouring the occurrence of translocations
To gain insight into the mechanisms of translocation formation that may lead to the clustering of breakpoints on chr 15, we searched for the presence of sequences known to promote recombination, and in consequence, rearrangements, within an approximate 2 Mb region surrounding the cluster of translocation breakpoints.
First, all the unbalanced translocations studied were found to have a breakpoint in the telomeric band of the partner chromosome which is known to contain potentially recombinogenic repetitive DNA sequences.24 Of the three translocations that have a breakpoint within the interstitial telomeric TTAGGG repeats, two also had a breakpoint within the 460 kb interval on15q (Table 1). This prompted us to search for perfect or degenerate TTAGGG telomeric repeats in the sequences of BACs located within the 2 Mb region, using a GCG programs (Accelrys)/SeqWeb interface. Nevertheless, we did not detect any significant concentration of telomere repeats in the sequence of these BACs.
Second the subtelomeric regions are also rich in various simple and low complexity repeats that are known to increase the recombination frequency.25 Using the Repeat masker Web Server, we observed that simple- and low-complexity repeats were both most frequent in BAC 323I15, which contains two translocation breakpoints (cases 1 and 8). In particular, in this BAC sequence, we found several (TA)n repeat tracts that can have nucleosome destabilizing properties which might promote greater chromatin accessibility, and in consequence increased recombination.26
Third telomeric bands of human chromosomes are known to be rich in members of the olfactory receptor (OR) gene family27 which may favour non-homologous recombination events with chromosome 15. However, we did not find any OR-related sequence in or near the 460 kb interval.
Lastly, several low copy repeat/LCR 15-duplicons, which are known to facilitate recombination events at meiosis and promote rearrangements, have been described on chromosome 15.18 Using the Human Genome Segmental Duplication Database Assembly program, we found a LCR15-duplicon element located adjacent to the BAC 64O8, very close to the proximal boundary of the 460 kb interval (Figure 4). This LCR15-duplicon (sequence AC027139) extends through 270 kb and is present at all other BPs that characterize recurrent rearrangements of chr 15.
Discussion
A new breakpoint cluster for recurrent rearrangements within the 15q14 band
We have demonstrated that the chr 15 breakpoint of four translocations with the telomeric band of the partner chromosome are clustered within a small 460 kb interval, located in the proximal 15q14 band. This interval is delineated by BACs 64O3 (proximal) and 150L8 (distal), and a LCR15-duplicon sequence is located adjacent to BAC 64O3. Such sequences have been suggested to lead to an open DNA/chromatin structure,28 favouring meiotic recombination and all types of chromosomal rearrangements. The presence in this chr 15 region of both a cluster of recurrent translocation breakpoints and an LCR15-duplicon sequence defines a new BP, which we named BP6. BP6 is more distal than all other BPs previously described in the proximal 15q region.
As for BP6, several previously described BPs characterize recurrent rearrangements of proximal 15q. Two proximal BPs have been defined based on either the presence (BP1) or the absence (BP2) of the D15S18 microsatellite marker within the aneusomic segment (Figure 1). BP1 and BP2 are involved in deletions, interstitial duplications and triplications with similar frequencies.4, 11, 12, 13, 29, 30, 31 They are also involved in small inv dup 15 chromosomes that are associated with a normal phenotype.7, 9, 32 Three distal BPs have been described (Figure 1). BP3 is the most common distal BP involved in PWS/AS deletions, interstitial duplications, and inv dup (15). It was initially located between markers D15S12 and D15S24 (33, for review) and has been subsequently more precisely located between markers D15S931 and D15S1019.12, 20, 31 Two other distal BPs, BP4 and BP5, which have not been fully characterized, are involved in cases of large 15q11–q14 interstitial triplications and inv dup (15) chromosomes. BP4 has been mapped between markers D15S1019 and D15S165 and BP5 between markers D15S165 and D15S144.10, 12, 18, 34
Indeed, a BP located distal to the marker D15S144 had previously been hypothesized,18, 34 but neither characterized nor delineated. Our results demonstrated that such a BP distal to BP5 exists and may be specific to translocations.
Is BP6 different from other breakpoint clusters?
Our study of eighteen large inv dup (15) chromosomes showed that none has a breakpoint distal to BP5, implying that none has a breakpoint within BP6. This result is particularly intriguing, and suggests that BP6 is different from the five other BPs which are all known to contain inv dup (15) breakpoints (34, for review).
Such a difference between BP6 and other proximal BPs is apparently not linked to the sequence itself. Indeed, our sequence analysis did not reveal any striking differences between BP6 and the other BPs. In particular, in the BP6 sequence, we did not find any concentration of telomere repeats that could favour translocations with the telomeric region of a partner chromosome. There is, therefore, no evidence that the mechanism of rearrangements involving BP6 is different from those involving the other BPs. Moreover, we demonstrate that BP6, like all five BPs described, previously18 contains LCR15-duplicon elements that are known to facilitate recombination events at meiosis and to promote rearrangements, either intrachromosomal35 or interchromosomal.36
One possible reason for the lack of inv dup (15) chromosomes having a breakpoint within BP6 could be the viability of the resulting phenotype. Indeed, rearrangements with a breakpoint in BP6 are associated with a segmental aneusomy larger than those with a more proximal breakpoint. Under such an hypothesis, inv dup (15) chromosomes (that lead to segmental tetrasomy 15q) would be associated with a less viable phenotype than unbalanced translocations (that lead to segmental monosomy 15q), when they have a breakpoint within BP6. To our knowledge, only one large inv dup (15) chromosome containing marker D15S144, the distal boundary of BP5, has been reported.34 However, this case has not yet been precisely characterized, molecularly or clinically, and therefore cannot contradict our hypothesis. Interestingly, the fact that the large monosomy 15q, resulting from unbalanced translocations involving BP6 are viable, even if they are infrequent, suggests that interstitial deletions delineated by breakpoints BP1/BP2 and BP6, should also exist.
Phenotype in cases of large monosomy 15q with a BP6 breakpoint
The large deletion of 15q resulting from unbalanced translocations with a breakpoint in BP6 is approximately twice as large as the interstitial deletions typically associated with type I (BP1-BP3) or type II (BP2-BP3) PWS/AS syndromes. The associated phenotype is therefore expected to be more severe or ‘expanded’ with respect to the typical PWS or AS phenotypes. The data from both the literature and our study clearly support an increased severity of the phenotype.
Rare unbalanced translocations have been reported in which a deleted segment that extends to cytogenetic bands 15q14 or 15q15, is larger than certain proximal 15q deletions, that extend to 15q13. Unfortunately, in the majority of cases, none of the breakpoints involved, either on chr 15q or on the recipient chromosome, has been precisely characterized and genotype–phenotype correlations remain difficult to establish.37, 38, 39, 40, 41, 42, 43, 44 Nevertheless, some translocations of this type have been shown to associate clinical signs of PWS, in addition to other findings not usually seen in this syndrome. This ‘expanded’ PWS phenotype may include cardiac, renal, and neurological abnormalities; bifida uvula and auditory dysfunction,42 has also been reported in two cases of large interstitial deletion of 15q,45, 46 indicating that it is linked to the loss of sequences from chr 15.
In our study, we demonstrated that cases 1, 3, 4, and 8 have an unbalanced translocation with the same BP6 breakpoint, and globally the associated phenotype is clearly more severe in these cases. Nevertheless, only Patient 1 presents some of the additional findings that characterize the ‘expanded’ PWS phenotype21 described by Schwartz et al.42 This difficulty in establishing precise genotype–phenotype correlations may have several explanations: the small number of cases analyzed, the parental origin of the rearrangement that leads to different phenotypes (PWS or AS), the fact that patients have been clinically described at different ages, the fact that some clinical signs have not been systematically investigated (auditory dysfunction and renal abnormality and so on), or finally a variable number of interstitial telomeric (TTAGGG)n repeats at the translocation breakpoint which could affect the expression of the adjacent genes by modifying the chromatin conformation at this site.
It should, however, be noted that the increased severity of the phenotype in our patients with a breakpoint within BP6, is not as ‘dramatic’ as would be expected from the extremely large size of the deletion (#10 Mb). This may be due to the relatively poor concentration of genes lying in this region of chr 15, and also to the fact that genes lying between BP3 and BP6 are not imprinted, and are therefore expressed from both alleles. In such a situation, deletion of one allele will decrease, but not suppress, the expression of the corresponding gene.
In conclusion, it should be very interesting to systematically delineate the boundaries of proximal 15q deletions, resulting from either unbalanced translocations or interstitial deletions. Indeed, it may be expected that some interstitial deletions have a breakpoint distal to BP3, which will allow the development of genotype–phenotype correlations. These correlations will aid in the identification of the genes that could be involved in the ‘expanded’ PWS or AS phenotypes, and will improve genetic counselling as well as clinical management for the patients. The DNA CGH-arrays technology with expanded coverage of the chr 15 sequences would be a well-adapted method for the accurate determination of deletion boundaries on the proximal chr 15q.
References
Knoll JH, Nicholls RD, Magenis RE, Graham Jr JM, Lalande M, Latt SA : Angelman and Prader–Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet 1989; 32: 285–290.
Robinson WP, Bottani A, Xie YG et al: Molecular, cytogenetic, and clinical investigations of Prader–Willi syndrome patients. Am J Hum Genet 1991; 49: 1219–1234.
Bundey S, Hardy C, Vickers S, Kilpatrick MW, Corbett JA : Duplication of the 15q11-13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol 1994; 36: 736–742.
Browne CE, Dennis NR, Maher E et al: Inherited interstitial duplications of proximal 15q: genotype–phenotype correlations. Am J Hum Genet 1997; 61: 1342–1352.
Cook Jr EH, Lindgren V, Leventhal BL et al: Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 1997; 60: 928–934.
Repetto GM, White LM, Bader PJ, Johnson D, Knoll JH : Interstitial duplications of chromosome region 15q11q13: clinical and molecular characterization. Am J Med Genet 1998; 79: 82–89.
Robinson WP, Binkert F, Gine R et al: Clinical and molecular analysis of five inv dup(15) patients. Eur J Hum Genet 1993; 1: 37–50.
Webb T : Inv dup(15) supernumerary marker chromosomes. J Med Genet 1994; 31: 585–594.
Mignon C, Malzac P, Moncla A et al: Clinical heterogeneity in 16 patients with inv dup 15 chromosome: cytogenetic and molecular studies, search for an imprinting effect. Eur J Hum Genet 1996; 4: 88–100.
Wandstrat AE, Schwartz S : Isolation and molecular analysis of inv dup(15) and construction of a physical map of a common breakpoint in order to elucidate their mechanism of formation. Chromosoma 2000; 109: 498–505.
Schinzel AA, Brecevic L, Bernasconi F et al: Intrachromosomal triplication of 15q11-q13. J Med Genet 1994; 31: 798–803.
Ungaro P, Christian SL, Fantes JA et al: Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J Med Genet 2001; 38: 26–34.
Vialard F, Mignon-Ravix C, Parain D et al: Mechanism of intrachromosomal triplications 15q11–q13: a new clinical report. Am J Med Genet 2003; 118: 229–234.
Rivera H, Zuffardi O, Gargantini L : Nonreciprocal and jumping translocations of 15q1-qter in Prader–Willi syndrome. Am J Med Genet 1990; 37: 311–317.
Jauch A, Robson L, Smith A : Investigations with fluorescence in situ hybridization (FISH) demonstrate loss of the telomeres on the reciprocal chromosome in three unbalanced translocations involving chromosome 15 in the Prader–Willi and Angelman syndromes. Hum Genet 1995; 96: 345–349.
Rossi E, Floridia G, Casali M et al: Types, stability, and phenotypic consequences of chromosome rearrangements leading to interstitial telomeric sequences. J Med Genet 1993; 30: 926–931.
Wenger SL, Sell SL, Painter MJ, Steele MW : Inherited unbalanced subtelomeric translocation in a child with 8p- and Angelman syndromes. Am J Med Genet 1997; 70: 150–154.
Pujana MA, Nadal M, Guitart M, Armengol L, Gratacos M, Estivill X : Human chromosome 15q11–q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet 2002; 10: 26–35.
Amos-Landgraf JM, Ji Y, Gottlieb W et al: Chromosome breakage in the Prader–Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 1999; 65: 370–386.
Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH : Large genomic duplicons map to sites of instability in the Prader–Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet 1999; 8: 1025–1037.
Windpassinger C, Petek E, Wagner K, Langmann A, Buiting K, Kroisel PM : Molecular characterization of a unique de novo 15q deletion associated with Prader–Willi syndrome and central visual impairment. Clin Genet 2003; 63: 297–302.
Krajewska-Walasek M, Gutkowska A, Bielinska B, Goryluk-Kozakiewicz B, Popowska E : A case of Prader–Willi syndrome arising as a result of familial unbalanced translocation t(11;15)(q25;q13). Clin Genet 1998; 54: 60–64.
Cuoco C, Bicocchi MP, Granata D, Mezzano P, Serra G : De novo (15;21) unbalanced translocation of paternal origin in a girl with Prader–Willi syndrome. Am J Med Genet 1990; 37: 62–64.
Flint J, Bates GP, Clark K et al: Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum Mol Genet 1997; 6: 1305–1313.
Murray J, Buard J, Neil DL et al: Comparative sequence analysis of human minisatellites showing meiotic repeat instability. Genome Res 1999; 9: 130–136.
Iyer V, Struhl K : Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J 1995; 14: 2570–2579.
Rouquier S, Taviaux S, Trask BJ et al: Distribution of olfactory receptor genes in the human genome. Nat Genet 1998; 18: 243–250.
Shaw CJ, Lupski JR : Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 2004; 13: R57–R64.
Knoll JH, Nicholls RD, Magenis RE et al: Angelman syndrome: three molecular classes identified with chromosome 15q11q13-specific DNA markers. Am J Hum Genet 1990; 47: 149–155.
Christian SL, Robinson WP, Huang B et al: Molecular characterization of two proximal deletion breakpoint regions in both Prader–Willi and Angelman syndrome patients. Am J Hum Genet 1995; 57: 40–48.
Roberts SE, Dennis NR, Browne CE et al: Characterisation of interstitial duplications and triplications of chromosome 15q11–q13. Hum Genet 2002; 110: 227–234.
Huang B, Crolla JA, Christian SL et al: Refined molecular characterization of the breakpoints in small inv dup(15) chromosomes. Hum Genet 1997; 99: 11–17.
Wandstrat AE, Leana-Cox J, Jenkins L, Schwartz S : Molecular cytogenetic evidence for a common breakpoint in the largest inverted duplications of chromosome 15. Am J Hum Genet 1998; 62: 925–936.
Robinson WP, Dutly F, Nicholls RD et al: The mechanisms involved in formation of deletions and duplications of 15q11–q13. J Med Genet 1998; 35: 130–136.
Ji Y, Eichler EE, Schwartz S, Nicholls RD : Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res 2000; 10: 597–610.
Spiteri E, Babcock M, Kashork CD et al: Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22 s) and telomeric bands of partner chromosomes. Hum Mol Genet 2003; 12: 1823–1837.
Borgaonkar DS, Ebenezer L, Scott Jr CI, Golomb HM, Bahr GF : Identification of a D–E(15–18) translocation chromosome by quinacrine fluorescence and urea banding techniques. Humangenetik 1973; 17: 317–321.
Ming PM, Goodner DM, Park TS : Chromosome 6/15 translocation with multiple congenital anomalies. Obstet Gynecol 1977; 49: 251–253.
Kawashima H, Fujita H, Sakamamoto Y, Hamamoto Y : An extra idic(15p)(q11) chromosome in Prader–Willi syndrome. Humangenet 1980, Cited as personal communication in: 55: 409–411.
Duckett DP, Roberts SH : Adjacent 2 meiotic disjunction. Report of a case resulting from a familial 13q;15q balanced reciprocal translocation and review of the literature. Hum Genet 1981; 58: 377–386.
Pauli RM, Meisner LF, Szmanda RJ : ‘Expanded’ Prader–Willi syndrome in a boy with an unusual 15q chromosome deletion. Am J Dis Child 1983; 137: 1087–1089.
Schwartz S, Max SR, Panny SR, Cohen MM : Deletions of proximal 15q and non-classical Prader–Willi syndrome phenotypes. Am J Med Genet 1985; 20: 255–263.
Smith A, Jauch A, St Heaps L, Robson L, Kearney B : Unbalanced translocation t(15;22) in ‘severe’ Prader–Willi syndrome. Ann Genet 2000; 43: 125–130.
Varela MC, Lopes GMP, Koiffmann CP : Prader–Willi syndrome with an unusually large 15q deletion due to an unbalanced translocation t(4;15). Ann Genet 2004; 47: 267–273.
Herva R, Vuorinen O : Congenital heart disease with del(15q) mosaicism. Clin Genet 1980; 17: 26–28.
Galan F, Aguilar MS, Gonzales J et al: Interstitial 15q deletion without a classic Prader–Willi syndrome. Am J Med Genet 1991; 38: 532–534.
Acknowledgements
The present work has been focused on unbalanced translocations involving chr 15q and the telomeric band of the partner chromosome. However, the authors would like to thank the members of the ACLF (Association des Cytogeneticiens de Langue Française) and the ECA (European Cytogeneticists Association) that kindly participate to this study by sending cytogenetic material: Drs G Bourrouillou (Toulouse, France); K Devriendt (Leuven, Belgique); A-M Frances (Toulon, France); B de Fréminville (St Etienne, France); E Gautier (Paris, France); H Journel (Vannes, France); N Joyé (Paris, France); P Kleinfinger (Cergy-Pontoise, France); J Lespinasse (Chambéry, France); D Martin-Coignard (Le Mans, France); K Miller (Hannover, Germany); H Moirot (Rouen, France); M Syrrou (Ionnanina, Greece); F Stipoljev (Zagreb, Croatie); H Stora de Novion (Nice, France); S Szpiro-Tapia (Cergy-Pontoise, France); L Taine (Bordeaux, France); and K Wagner (Graz, Austria). This work was supported by grants from the ARC (Association pour la Recherche contre le Cancer), INSERM and the Ministère de l'Enseignement et de la Recherche.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mignon-Ravix, C., Depetris, D., Luciani, J. et al. Recurrent rearrangements in the proximal 15q11–q14 region: a new breakpoint cluster specific to unbalanced translocations. Eur J Hum Genet 15, 432–440 (2007). https://doi.org/10.1038/sj.ejhg.5201775
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201775
Keywords
This article is cited by
-
Prader-Willi syndrome - type 1 deletion, a consequence of an unbalanced translocation of chromosomes 13 and 15, easily to be mixed up with a Robertsonian translocation
Molecular Cytogenetics (2015)
-
A study of two Chinese patients with tetrasomy and pentasomy 15q11q13 including Prader-Willi/Angelman syndrome critical region present with developmental delays and mental impairment
BMC Medical Genetics (2013)
-
Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes
European Journal of Human Genetics (2012)
-
Copy Number Variants: A New Molecular Frontier in Clinical Psychiatry
Current Psychiatry Reports (2011)
