
Abstract
A large multiplex family presumably affected with autosomal recessive cone–rod dystrophy (CRD) was ascertained from Israel. In this family of Christian Arab ancestry with six consanguineous loops, linkage analysis failed to identify homozygosity in all six nuclear families at any of the three arCORD loci hitherto reported. However, homozygosity was found at the CORD3 locus for two nuclear families and the segregation of three distinct haplotypes at this locus in the whole pedigree suggested the alteration of the ABCA4 gene. This hypothesis was confirmed by the identification of three distinct mutations. Subsequently, with regard to the wide spectrum of autosomal recessive retinal dystrophies related to ABCA4 mutations, the natural history of the disease was revisited in all patients. Although the diagnosis of CRD was confirmed in 8/9 patients, the last one, aged of 34, displayed typical signs of Stargardt disease without extension to the peripheral retina. The results of this study emphasize the pitfalls of homozygosity mapping in highly inbred families when the heterozygote carrier frequency is particularly high in the general population.
Similar content being viewed by others


Introduction
The ABCA4 gene1, 2 has been associated with several forms of inherited autosomal recessive retinal dystrophies (MIM 601691). Indeed, mutations in this retinal-specific ATP-binding cassette gene account for all cases of early- and late-onset autosomal recessive Stargardt disease (arSTGD; MIM 248200) and for the main part of autosomal recessive cone–rod dystrophies (arCRDs).3, 4 Some mutations in ABCA4 have been reported in exceptional cases of retinitis pigmentosa (RP).5, 6
A substantial allelic heterogeneity has been described (Human Gene Mutation Database). In addition, the carrier frequency of ABCA4 variants is particularly high in the general population, ranging from 1/50 to 1/10.7, 8, 9
Within the huge family of inherited retinal dystrophies, the CRD phenotype indicates a specific form of retinal degeneration in which the cone degeneration appears early in life with a central involvement of the retina, followed by the degeneration of rods several years later.9 This particular form of retinal dystrophy has been regarded as ‘inverse RP’ and can be misdiagnosed as macular dystrophy in the first stages of the disease. Indeed, the main symptoms of arCRD at disease onset are decrease of visual acuity, loss of color discrimination and photophobia. The b-wave of the photopic electroretinogram (ERG; cone response) is severely reduced, although the b-wave of the scotopic ERG is still normal. As the disease progresses, nyctalopia, progressive peripheral visual field deficit and decreasing scotopic ERG amplitudes are observed.
Here, we report the identification of three ABCA4 disease alleles in a large multiplex family from Israel with six consanguineous loops gathering nine patients affected with autosomal recessive retinal dystrophy. This study confirms that, in view of the high frequency of ABCA4 mutation carriers, the existence of more than one disease-causing mutation should be considered in consanguineous families.
Patients and methods
Patients
A large multiplex family of Christian Arab ancestry affected with autosomal recessive retinal dystrophy was ascertained through the Genetic Institute of Ha’Emek Medical Center in Afula, Israel. This pedigree was made of six loops of consanguinity gathering nine affected members and seven healthy relatives.
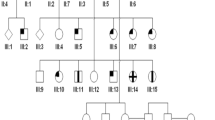
In a first step, individuals of only three of the six loops of the pedigree (L1–L3; Figure 1) were addressed to the Laboratory. All affected patients of these three loops (III1, III2, IV1, IV2, IV4) underwent general and ophthalmologic examinations in Afula. All of them fulfilled the minimal criteria for the diagnosis of CRD10.

Pedigree of a Christian Arab family of Israel and haplotypes at the CORD3 locus on chromosome 1p22.1. Three haplotypes segregate with the disease, each carrying a distinct ABCA4 mutation. M1: c.5460+1G>A; M2:p.G1961E and M3:p.G1202R. The six loops of consanguinity of this pedigree are labelled L1–L6, respectively.
In a second step, family members of three additional loops of the pedigree (L4–L6; Figure 1), gathering four individuals considered as affected with the same disease, were addressed to the laboratory for genetic studies. Precise clinical data were requested for these last patients and obtained later on.
Each family member's blood was collected in Afula and analyzed in France.
Linkage analysis at arCRD loci
A linkage analysis was performed in this pedigree using markers containing short tracks of (CA)n repeats flanking each of the three hitherto known arCRD loci: AFMa051wg9 (D1S2868), AFM350tg9 (D1S2849) and AFM205ta11 (D1S236) at the CORD3 locus on chromosome 1p22 (MIM 604116); AFMa297 × g9 (D1S2696), AFM234zd12 (D1S442), AFMa046wd9 (D1S2343), AFMa244wh5 (D1S2675) and AFM350yh1 (D1S2851) at the CORD8 locus on chromosome 1q12q24 (MIM 605549); and AFMb329we5 (D14S1023) and AFM312xh1 (D14S283) at the CORD9 locus on chromosome 11q11 (MIM 608194), respectively. All nucleotide sequences, allelic frequencies and distances between markers were available from Genethon linkage map11 and the Human Gene Mutation Database.
In addition, intronic primers were designed to flank short tracks of nucleotide repeats in the ABCA4 and RPGRIP1 genes at the CORD3 and CORD9 loci, respectively. The nucleotide sequences of RPGRIP1 primers were available elsewhere.12 ABCA4 primer sequences were: forward 5′TTAAGAGAACTTATTTGGCATAGGA3′; reverse 5′CAAAGAACCGGAAGAGACTGT3′.
Amplified fragments were electrophorezed and analyzed on an automatic sequencer (ABI3130, Applied Biosystems, Foster City, CA, USA).
Mutational screening of the ABCA4 gene
All 50 exons of the ABCA4 gene were PCR amplified and subsequently sequenced as described previously.13
Mutation nomenclature
We have chosen to number the A of the start codon (ATG) of the GenBank ABCA4 cDNA sequence (NM_000350) as nucleotide +1.
Results
A Segregation analysis was performed in the three first loops of the pedigree using at least four polymorphic markers flanking each arCRD locus (L1–L3; Figure 1). No homozygosity was found for all five affected members at any of the three arCRD loci (available on request). However, the affected sibs of one of the three nuclear families were found to be homozygous at the CORD3 locus (Figure 1). Considering the high ABCA4 carrier frequency, we made the hypothesis of the involvement of this gene at the CORD3 locus in this family, assuming that three distinct ABCA4 disease haplotypes might exist. The genetic study of the three additional loops of this pedigree was consistent with this hypothesis.
Three different ABCA4 mutations were identified: (i) c.5460+1G>A (M1), (ii) c.5882G>A (p.G1961E; M2) and (iii) c.3607G>A (p.G1202R; M3). Among these three mutations only one was novel, c.3607G>A (p.G1202R; M3). Two out of the 143 control individuals of our series were shown to be heterozygous for this mutation (2/286 chromosomes; 0.7%).
Patients IV1-IV2-IV7-IV8 were homozygous for the c.5460+1G>A splice-site mutation (M1), whereas patients III1–III2 and IV4-V1 were compound heterozygous for this mutation (M1) and the c.5882G>A (M2) or c.3607G>A (M3) missense mutations, respectively. Finally, patient IV5 was compound heterozygous for these last two missense mutations (M2–M3; Figure 1).
The natural history of the disease and the ophthalmologic data were revisited in all nine affected patients of the pedigree. The diagnosis of CRD was confirmed in patients III1, III2, IV1, IV2, IV4, IV7, IV8 and V1, as they displayed the minimal criteria for this diagnosis described elsewhere. The last patient IV5, aged of 34, displayed typical signs of Stargardt disease without extension to the peripheral retinal. ERG recordings showed severely reduced photopic responses, whereas scotopic ERG amplitudes were normal.
The three mutations identified were found to segregate with the disease in the family (Figure 1) with the exception of a child aged of 3 (V3) who harbored two presumed disease haplotypes, but whose clinical status could not be determined considering her age.
Discussion
Mutations of the ABCA4 gene have been reported to account for 30–80% of arCRDs.3, 4 In addition, it is admitted that the frequency of carriers of ABCA4 mutations in the general population ranges from 1/20 to 1/10.7, 8, 9 In light of these data, we made the hypothesis of the localization of the disease-causing gene in this inbred family at the CORD3 locus despite the absence of homozygosity for all patients. This hypothesis was supported by the existence of one common haplotype identified in all affected members of the three first loops studied in a first step (M1; Figure 1). The genetic study extended to three additional loops was consistent with the existence of three distinct ABCA4 disease haplotypes. However, whereas 3/4 affected members of these additional loops harbored the M1 haplotype (IV7, IV8, V1; Figure 1), the last one IV5 carried the two other haplotypes (M2 and M3; Figure 1). The evidence of three disease haplotypes was confirmed by the identification of three different ABCA4 mutations: one splice site mutation and two missense mutations carried by the M1, M2 and M3 haplotypes, respectively. Interestingly, 5/9 patients born to consanguineous parents belonging to 4/6 loops of consanguinity were found to be compound heterozygous for two of the three mutations. Among these five patients, three different haplotype combinations were found: M1M2, M1M3 and M2M3 (Loops L1, L3–L5 and L4, respectively). On the other hand, homozygosity for M1 was identified in 4/9 patients belonging to the L2 and L6 consanguineous loops (Figure 1). Considering the previously reported ABCA4 genotype–phenotype correlations14, 15, 16 and the existence of four different combinations of ABCA4 mutations, the natural history of the disease and the ophthalmologic data were revisited for all patients.
The four affected members of this pedigree homozygous for ABCA4 splice-site mutation: c.5460+1G>A (M1; patients IV1, IV2, IV7, IV8) were severely affected since the first decade of life (cone dysfunction followed by rod dysfunction in the end of the second decade).
The two affected sibs compound heterozygote for the c.5460+1G>A splice-site mutation (M1) and the p.G1961E mutation (M2; patients III1, III2) were as severely affected as patients homozygous for the splice-site mutation. Thus, it is likely that the p.G1961E mutation significantly affects the function of the protein. This notion is supported by the functional study of this mutation, which showed that the mutant protein exhibited a reduced basal ATPase activity that is inhibited, rather than stimulated, by retinal.17 Additionally, it is worth noting that the p.G1961E mutation has already been found to be associated with arCRD.18, 19
Two patients were compound heterozygous for the c.5460+1G>A splice-site mutation (M1) and the p.G1202R substitution (M3, patients IV4 and V1; Figure 1). Interestingly, this last change was found in a heterozygous state in 2/143 control individuals of the general population (1/70). This frequency is higher than expected with regard to the heterozygous frequency of ABCA4 mutation in the general population (1/50–1/10).7, 8, 9 However, it is worth noting that this change has never been reported previously, neither in the Stargardt population nor in the general population suggesting that its identification in two individuals in our multiethnic control population may be random. In addition, the glycine at position 1202 lies in a conserved region and is highly conserved in mice, dogs, bulls and frogs. In addition, the mutation changes a nonpolar aliphatic amino acid into a charged amino acid. One out of the three patients (IV4) developed a retinal degeneration as early as 7 years. The disease confined in early stages to the macular region extended to the peripheral retina by the age of 20 years as shown by decreased ERG responses for both scotopic and photopic components. The second patient (V1), aged of 10, displays typical signs of Stargardt disease with decrease of central visual acuity, dark choroid and fundus flavimaculatus at the fluorescein angiography. Nevertheless, the age of this girl does not allow prediction as to the final prognosis of the disease, which in this family, progresses to a rod dysfunction characterized by night blindness, reduction of the visual field and pigmentary deposits in the peripheral retina at the end of the second decade. The last patient (V3), aged of 3, was found to be haploidentical to her elder sister (V1) but is too young to present symptoms of the disease.
Finally, one patient (IV5) was found to carry the p.G1961E and p.G1202R missense mutations. This patient, mother of V1 and V3, aged of 34 and considered as affected with the same disorder than her relative was in fact affected with a typical STGD disease, that is, without extension of the disease to the peripheral retina. The phenotype of this woman associated with compound heterozygosity for the p.G1961E and p.G1202R mutations is less severe than the association of one of them with the c.5460+1G>A splice-site mutation. These findings are consistent with the previously reported ABCA4 genotype–phenotype correlations, on one hand,15, 20, 21 and with the report of families segregating different phenotypes associated with different ABCA4 mutations, on the other hand.6, 16, 22, 23, 24, 25
This study emphasizes the homozygosity pitfals in highly inbred families when the heterozygote carrier frequency is known to be much higher than the mean frequency of heterozygote individuals in the general population.
References
Allikmets R, Singh N, Sun H et al: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997; 15: 236–246.
Gerber S, Rozet JM, van de Pol TJ et al: Complete exon–intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease. Genomics 1998; 48: 139–142.
Maugeri A, Klevering BJ, Rohrschneider K et al: Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone–rod dystrophy. Am J Hum Genet 2000; 67: 960–966.
Ducroq D, Rozet JM, Gerber S et al: The ABCA4 gene in autosomal recessive cone–rod dystrophies. Am J Hum Genet 2002; 71: 1480–1482.
Martinez-Mir A, Paloma E, Allikmets R et al: Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998; 18: 11–12.
Rozet JM, Gerber S, Ghazi I et al: Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet 1999a; 36: 447–451.
Maugeri A, Flothmann K, Hemmrich N et al: The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur J Hum Genet 2002; 10: 197–203.
Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR : Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet 2001; 108: 346–355.
Jaakson K, Zernant J, Kulm M et al: Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003; 22: 395–403.
Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K : Phenotypic spectrum of autosomal recessive cone–rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci 2002; 43: 1980–1985.
Dib C, Faure S, Fizames C et al: A comprehensive genetic map of the human genome based on 5264 microsatellites. Nature 1996; 380: 152–154.
Hanein S, Perrault I, Gerber S et al: Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23: 306–317.
Gerber S, Rozet JM, van de Pol TJ et al: Complete exon–intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease. Genomics 1998; 48: 139–142.
Rozet JM, Gerber S, Souied E et al: The ABCR gene: a major disease gene in macular and peripheral retinal degenerations with onset from early childhood to the elderly. Mol Genet Metab 1999b; 68: 310–315.
Lewis RA, Shroyer NF, Singh N et al: Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet 1999; 64: 422–434.
Cremers FP, van de Pol DJ, van Driel M et al: Autosomal recessive retinitis pigmentosa and cone–rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet 1998; 7: 355–362.
Sun H, Smallwood PM, Nathans J : Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet 2000; 26: 242–246.
Klevering BJ, Yzer S, Rohrschneider K et al: Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone–rod dystrophy and retinitis pigmentosa. Eur J Hum Genet 2004; 12: 1024–1032.
Rudolph G, Kalpadakis P, Haritoglou C, Rivera A, Weber BH : Mutations in the ABCA4 gene in a family with Stargardt's disease and retinitis pigmentosa (STGD1/RP19). Klin Monatsbl Augenheilkd 2002; 219: 590–596.
Maugeri A, van Driel MA, van de Pol DJ et al: The 2588G → C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 1999; 64: 1024–1035.
Rozet JM, Gerber S, Souied E et al: Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet 1998; 6: 291–295. (Erratum in: Eur J Hum Genet 1999; 7: 102).
Ozdek S, Onaran Z, Gurelik G, Bilgihan K, Acar C, Hasanreisoglu B : Stargardt's disease and retinitis pigmentosa: different phenotypic presentations in the same family. Eye 2005; 19: 1222–1225.
Paloma E, Coco R, Martinez-Mir A, Vilageliu L, Balcells S, Gonzalez-Duarte R : Analysis of ABCA4 in mixed Spanish families segregating different retinal dystrophies. Hum Mutat 2002; 20: 476.
Rudolph G, Kalpadakis P, Haritoglou C, Rivera A, Weber BH : Mutations in the ABCA4 gene in a family with Stargardt's disease and retinitis pigmentosa (STGD1/RP19)]. Klin Monatsbl Augenheilkd 2002; 219: 590–596, German.
Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR : Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 2001; 42: 2757–2761.
Author information
Authors and Affiliations
Corresponding author
Additional information
Electronic database information ABCA4, MIM601695, Genbank accession number NM_000350
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/entrez/
UCSC Human Genome Project working draft, http://genome.ucsc.edu (for marker order and genetic distance between markers).
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/hgmd0.html
Rights and permissions
About this article
Cite this article
Ducroq, D., Shalev, S., Habib, A. et al. Three different ABCA4 mutations in the same large family with several consanguineous loops affected with autosomal recessive cone–rod dystrophy. Eur J Hum Genet 14, 1269–1273 (2006). https://doi.org/10.1038/sj.ejhg.5201691
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201691
Keywords
This article is cited by
-
The molecular basis of autosomal recessive diseases among the Arabs and Druze in Israel
Human Genetics (2010)
