
Abstract
In the majority of patients with a chromosome 15 imprinting defect (ID) causing Prader–Willi syndrome (PWS) or Angelman syndrome (AS), the defect is a primary epimutation that occurred spontaneously in the absence of a DNA mutation. We have investigated whether common DNA sequence variants in the bipartite imprinting centre (IC) are associated with an increased susceptibility to imprinting defects. We have determined the haplotype structure of the IC and found that the two IC elements called ‘PWS-SRO’ and ‘AS-SRO’ lie on separate haplotype blocks. To identify susceptible IC sequence variants, we have used the transmission disequilibrium test. While we did not observe preferential transmission of a paternal allele or haplotype in 41 PWS-ID trios, we found a trend for preferential maternal transmission of one AS-SRO haplotype (H-AS3) in 48 AS-ID trios (P=0.058) and could identify two sequence variants in H-AS3 that are responsible for this effect. We also obtained tentative evidence that homozygosity for the 677C>T variant of the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene on chromosome 1 might increase the risk of a maternal imprinting defect: the frequency of the TT genotype was significantly higher in the mothers of the AS patients with an imprinting defect than in the patients' fathers or the general population (P=0.028). Our findings suggest that women with the IC haplotype H-AS3 or homozygosity for the MTHFR 677C>T variant may have an increased risk of conceiving a child with an imprinting defect, although the absolute risk is low.
Similar content being viewed by others

Introduction
Prader–Willi syndrome (PWS) and Angelman syndrome (AS) are distinct neurogenetic disorders caused by the loss of function of imprinted genes in proximal 15q.1 Most of the patients have a large deletion of 15q11–q13, uniparental disomy 15, or, in AS, a UBE3A mutation. In 1% of patients with PWS and in 3% of patients with AS the disease is due to an imprinting defect (ID). In patients with PWS and an ID, the paternal chromosome carries a maternal imprint, whereas in patients with AS and an imprinting defect, the maternal chromosome carries a paternal imprint. A microdeletion affecting the imprinting centre (IC) accounts for 10–15% of imprinting defects.2 In the vast majority of patients with an imprinting defect, however, the defect is a primary epimutation that occurred spontaneously in the absence of an IC mutation.3
The IC overlaps the SNURF-SNRPN locus and has a bipartite structure. One element is defined by deletions in patients with PWS and an imprinting defect. The smallest region of deletion overlap in these patients, the PWS-SRO, is 4.1 kb in size (Ohta et al4 and personal communication of H.-J. Dong and D. Driscoll) and includes the SNURF-SNRPN promoter and exon 1. The deletions impair the postzygotic maintenance of the paternal imprint.5 The second element is defined by deletions in patients with AS and an ID. The smallest region of deletion overlap in these patients (the AS-SRO) comprises 880 bp and maps 35 kb upstream of exon 1 of SNURF-SNRPN. The deletions impair the establishment of the maternal imprint in the female germ line.2 There is some evidence that in this process the AS-SRO element interacts with the PWS-SRO element.2, 6, 7, 8 The molecular mechanisms are unknown, but likely to involve trans-acting factors that bind to the AS-SRO and the PWS-SRO. Therefore, it is possible that DNA sequence variations in the protein binding sites of the IC might affect the binding of trans-acting factors and consequently the epigenetic state of the chromosomal domain. In fact, there is some evidence that common DNA sequence variants may be associated with an increased risk of loss of imprinting: Sandovici et al9 found familial aggregation of abnormal methylation of parental alleles at the IGF2/H19 and IGF2R differentially methylated regions and concluded that genetic factors are responsible for a major fraction of inter-individual variability in parental origin-dependent epigenetic modifications. Additionally, as shown by Murrel et al,10 some variants of the IGF2 gene may be associated with Beckwith–Wiedemann syndrome. The association was still significant in a BWS subgroup with loss of methylation at the KvDMR1, which account for approximately 50% of patients. As the authors did not determine the haplotype structure of the 500 kb IGF2-KvDMR1/LIT1 region, it is unclear whether the IGF2 variants are in linkage disequilibrium with KvDMR1/LIT1, or whether there are long-range interactions. Also, the authors used a simple case–control study, which is sensitive to population structure. Independent studies on this topic are therefore necessary to clarify the mechanism of interaction of allelic effects and IDs.
It is possible that the spontaneous epimutation rate is also modified by trans-acting genetic factors. Direct trans-acting factors that bind to the AS-SRO and the PWS-SRO are not known so far, but a good candidate for an indirect trans-acting factor is the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene. MTHFR is a key regulatory enzyme in the one-carbon metabolism and plays an important role in folate metabolism, DNA methylation and DNA synthesis. It catalyzes the reduction of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, which is used by the methionine synthase for the methylation of homocysteine to methionine. Methionine is the precursor of S-adenosyl-L-methionine (SAM), which serves as a methyl donor for DNA and histone methyltransferases. A common 677C>T transition in the MTHFR gene results in a thermolabile protein with reduced enzymatic activity.11 Changes in MTHFR activity affect the levels of SAM and several studies have shown that homozygosity for the T allele is associated with global genomic DNA hypomethylation.12, 13, 14, 15 Although imprints appear to be buffered against global changes in DNA methylation,16 it is possible that reduced levels of SAM in female germ cells increase the risk that maternal methylation imprints are not or not completely established.
Over the past 10 years we have established a unique collection of DNA samples from PWS and AS patients with an imprinting defect. Here we report that common DNA sequence variants of the 15q imprinting centre and the MTHFR gene are associated with an increased tendency of epimutations in the PWS/AS region.
Methods
Patients
All patients were diagnosed by experienced clinicians. In patients with an imprinting defect, biparental inheritance of the PWS/AS region was shown by microsatellite analysis. Methylation analysis of SNURF-SNRPN promoter/exon 1 region by MS-PCR17 revealed that the PWS patients lack an unmethylated allele and that the AS patients lack a methylated allele. An IC deletion has been excluded in all these patients. Demographic data and sex of the patients are given in Supplementary Table 1a and b.
To determine the haplotype structure of the IC we studied DNA samples from patients with a de novo deletion 15q11–q13 and selected parental samples. The deletion was diagnosed either by FISH or microsatellite analysis.
The study was approved by the local ethics committee.
SNP genotyping
For genotyping SNPs rs2355600 (P7), rs220030 (P8) and rs220028 (P9) we used a TaqMan 5′-allele discrimination Assay-by-Design method (Applied Biosystems, Darmstadt, Germany). The sequences for the primers and probes used for the three assays are as follows:
P7: 5′-GTTTTGTTTATTCTTAACTGTATGTATTTTTTTGTCTTTTCC-3′ (forward primer), 5′-GAACACCATCAATAAAAATTAAGAAGCTGGAA-3′ (reverse primer), 5′-CATTTTCTGTATTTATCC-3′ (minor groove binder) and 5′-ATTTTCTGTGTTTATCC-3′ (minor groove binder).
P8: 5′-ATTGATTGTGGTTATGGCGCATTT-3′ (forward primer), 5′-CTCACCCTCAGGTCTTCCTATGT-3′ (reverse primer), 5′-CCAGCTTTTTTGTACCGC-3′ (minor groove binder) and 5′-CAGCTTTTTCGTACCGC-3′ (minor groove binder).
P9: 5′-GGTGGGCATGGCATGGA-3′ (forward primer), 5′-AGCAATCATTCAGCGTCCCTTAG-3′ (reverse primer), 5′-TCACTCCGGACAGCG-3′ (minor groove binder) and 5′-CACTCCAGACAGCG-3′ (minor groove binder).
For the 677C>T mutation (rs1801133) in the MTHFR gene we used for TaqMan 5′ allele discrimination an Assay-on-Demand (Applied Biosystems, Assay-ID: C___1202883_20). The probes were labelled with the fluorescent dyes VIC and FAM, respectively. The polymerase chain reaction was carried out with 12.5 ng DNA in a final reaction volume of 25 μl using the following amplification conditions: denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 92°C for 15 s and annealing and extension at 60°C for 1 minute (except for P8, where the annealing and extension temperature was 62°C). The samples were analyzed on an ABI PRISM Sequence Detection System 7000, using the SDS 1.1 software for allelic discrimination (Applied Biosystems).
The remaining polymorphisms P1 (rs2075814), P2 (rs2736711), P3 (ss49785042), P4 (ss49785041), P5 (rs17114852), P6 (rs17785249) and P10 (rs4906940) were studied by sequence analysis. The AS-SRO was analyzed by amplifying a 1184 bp PCR product with primers IC16 and MOP3 and sequencing of the PCR products with the same primers. For P10 in the SNURF-SNRPN intron 1 region, primers SNRPNi1 and SNRPNi2 were used to amplify a 409 bp PCR product and primer SNRPNi2 was used for the sequencing reaction. The primer sequences have been previously described.3 PCR products were purified with the MultiScreen Filtration System (Millipore, Schwalbach, Germany). Sequencing reactions were performed with fluorescence-tagged dideoxynucleotides (BIGDye kit) and the Taq cycle sequencing procedure (Applied Biosystems, Darmstadt, Germany). Sequences were analyzed on an ABI 3100 DNA Sequencer.
Haplotypes
Linkage disequilibrium (LD) block structure was examined using the software Haploview, version 2.05.18 This software provides computation of LD statistics and population haplotype patterns from primary genotype data. Pairwise LD between polymorphisms is expressed as D′ (a normalized measure for assessing the strength of LD). Haplotype blocks were generated using the 4-gamete rule algorithm.
The relationship between the haplotypes of the AS-SRO and the relationship between the haplotypes of the PWS-SRO was examined by a median-joining approach,19 using the software Network 4.1.1.0 (http://www.fluxus-engineering.com).
Statistical analysis
All analyses were performed using the R software package (http://www.r-project.org). To conduct the multiallelic TDT (mTDT) we used the package gap which implements Sham.20 The mTDT was used to determine association of haplotypes. In the trio sample phases were resolved using family information and eliminating remaining ambiguities by restricting possible haplotypes to those characterized in the deletion patients. The biallelic TDT was performed as described by Spielman et al.21 All 2 × 2 contingency tables were tested using Fisher's exact test. 3 × 2 contingency tables were evaluated using Pearson's χ2-test. All loci were subjected to a goodness-of-fit test for Hardy–Weinberg equilibrium (HWE). None of the loci showed departure from HWE (P>0.4).
Results
IC sequence variants
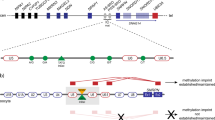
By sequence analysis of the critical IC elements we had previously identified four single nucleotide polymorphisms (SNPs) and one 4-bp insertion/deletion polymorphism in the AS-SRO (P1–P5), one SNP 9 bp distal to the AS-SRO (P6), and two SNPs close but distal to the PWS-SRO (P9, P10).3, 22 P9 is already known as an MspI polymorphism.22 By data base searches we identified another two SNPs: one SNP 1.4 kb distal to the AS-SRO (P7) and one SNP in the PWS-SRO (P8). The location of the ten polymorphisms is shown in Figure 1. All polymorphisms have a minor allele frequency >0.06.

Physical map of the IC region. The positions of the polymorphisms are indicated by vertical bars. In the text, they are refered to as P1–P10 (from centromere to telomere). Black boxes indicate the SNURF-SNRPN exons. Polymorphisms that were analyzed with Assays-by-Design are indicated by a diamond. Polymorphisms that were analyzed by sequence analysis are indicated by an asterisk.
Identification of IC haplotypes
First we determined the haplotype structure of the IC region. As PWS and AS patients with a large de novo deletion of 15q11–q13 have only one copy of this region, we are in the unique position to define the haplotypes experimentally. In order to increase the number of haplotypes we have determined the non-transmitted haplotype by comparing the haplotype of each PWS deletion patient with the genotype of the mother and the haplotype of each AS deletion patient with the genotype of the father. We studied the IC polymorphisms in 110 patients with PWS and 16 mothers and in 71 patients with AS and 65 fathers. By this, a total of 262 haplotypes could be observed.
Using the Haploview software we identified two blocks of LD (Figure 2). One block spans the polymorphisms P1–P6; the other block spans P8–P10. P7 is between the two blocks. We also analyzed the data generated by the International Hap Map Project. Of the 10 polymorphisms studied by us, only P1, P7 and P8 are included in the Hap Map Project. The Hap Map data show that the PWS-SRO is part of a larger haplotype block spanning the 5′ end of SNURF-SNRPN, whereas linkage disequilibrium in the AS-SRO is confined to this element (data not shown). Consequently, we analyzed both IC elements separately.

Haplotype structure of the IC as analyzed with Haploview. Each square represents a pairwise comparison between two polymorphisms (P1–P10). Values for D′ (× 100) are shown in the squares. Bright red squares indicate pairwise D′ values=1 and a statistically significant LD between the pair of polymorphisms. Blue squares also indicate pairwise D′ values=1 but without statistical significance. Squares shaded pink or red and white squares indicate pairwise D′ values <1, pink or red squares with statistical significance and white squares without statistical significance. The two haplotype blocks are outlined as triangular regions.
Haplotypes of the AS-SRO
For the AS-SRO we observed six different haplotypes (Table 1), which occur at a frequency of 0.49, 0.23, 0.13, 0.08, 0.06 and 0.004, respectively. Using the Fluxus software we investigated the degree of relationship between the haplotypes (Figure 3a). The second and third frequent haplotypes H-AS2 and H-AS3 differ in two polymorphisms from the most frequent haplotype H-AS1. The three rare haplotypes H-AS4, H-AS5 and H-AS6 differ in only one polymorphism from haplotype H-AS1.

Median-joining network of the haplotypes. (a) AS-SRO. (b) PWS-SRO. Each circle represents one haplotype. The area of each circle is proportional to the frequency of the haplotype. The haplotypes differ in sequence at the polymorphic sites indicated next to the links.
Haplotypes of the PWS-SRO
For the PWS-SRO we observed four different haplotypes, two frequent and two rare haplotypes (Table 2). Haplotype H-PWS1 occurred at a frequency of 0.6 and haplotype H-PWS2 at a frequency of 0.35. The frequencies of the two rare haplotypes H-PWS3 and H-PWS4 are 0.04 and 0.004. The two frequent haplotypes differ in all three polymorphisms. The two rare haplotypes differ in only one polymorphism from the most frequent haplotype (Figure 3b).
Association studies with IC sequence variants
In order to identify IC sequence variants with an increased susceptibility to imprinting defects, we genotyped a cohort of 48 AS-ID trios (affected child, mother and father) and 41 PWS-ID trios. To test for association, we used the transmission disequilibrium test (TDT). The TDT is a family-based association test that determines whether an allele or haplotype is transmitted from heterozygous parents to affected offspring more often than it would be expected by chance, that is on more than 50% of occasions. One advantage of the TDT is that it is robust against population structure, which is relevant for the current study, because the families have been recruited worldwide. Furthermore, a family-based test easily allows to take into account whether the affected haplotype is inherited from the father or from the mother. We calculated a biallelic TDT for the haplotypes and a multiallelic TDT for single polymorphisms and combinations of the polymorphisms. In the PWS-ID trios we did not observe a preferential transmission of an allele or haplotype (data not shown). In the AS-ID trios we observed that the AS-SRO haplotype H-AS3 was transmitted by the mothers more often than expected (P=0.058); (Table 3). A multiallelic TDT revealed a trend for an increased transmission of allele G of P1 and the 4 bp deletion of P3 and the combination of these polymorphisms (P=0.058); (Table 4). Furthermore, we obtained significant P-values (P=0.021) for three combinations of P1, P3 and P4. Over-transmission was detected for allele G of P1, for the 4 bp deletion of P3 and for allele C of P4. Of note, allele G of P1 and the 4 bp deletion allele of P3 are unique to haplotype H-AS3 and not found in chimpanzee, gorilla, orang-utan, macaque, baboon and wooly monkey (R.D. Nicholls and K. Yamasaki, personal communication).
In the AS patients, we observed only two rare variants in the AS-SRO, both on haplotype H-AS1. One variant (T insertion bp 6525–65263) was inherited from the patient's father. Another one (6048 C/G [GenBank accession number AF148319]) was inherited from the patient's mother. Both variants are outside functional AS-SRO elements as defined by Kantor et al.23
Analysis of MTHFR variants
To determine whether homozygosity for the MTHFR 677 T allele affects the establishment of the maternal imprint, we genotyped 52 mothers of AS patients with an imprinting defect. As the trios were recruited worldwide, we used the fathers as controls, because they are likely to belong to the same ethnic groups as the mothers. The distribution of the genotypes of 52 mothers and 50 fathers of AS-ID patients is shown in Table 5. Using Fisher's exact test we revealed that the frequency of the 677TT genotype is significantly increased in mothers (P=0.028). The odds ratio is 5.71 with a 95% confidence interval (CI) of 1.18–27.56 (Pearson's χ2 for a 3 × 2 contingency: χ2=7.76, df=2, P=0.021). The use of the fathers' genotypes as controls is only valid, if the paternal genotype does not affect the risk of an epimutation on the maternal allele. Although we consider this scenario unlikely, we also calculated the odds ratio and the P-value using the genotype frequencies determined by van der Put et al.24 The genotype frequencies in this population are similar to the frequencies in our group of fathers. Using these data, we obtained an odds ratio of 2.43 with a 95% CI of 1.12–5.24 and a P-value of 0.028 (Pearson's χ2 for a 3x2 contingency: χ2=6.94, df=2, P=0.031).
Discussion
Imprinting defects are epimutations that result from errors in imprint erasure in primordial germ cells, imprint establishment during later stages of gametogenesis, or imprint maintenance after fertilization. In the majority of AS and PWS patients with an imprinting defect, the errors are not caused by an imprinting centre mutation, but probably represent stochastic events. Here we provide tentative evidence that the rate of certain errors can be increased by cis- and trans-acting genetic factors.
Our data suggest that the AS-SRO haplotype H-AS3 might be associated with an increased risk of an imprinting defect on the maternal chromosome. Analysis of the individual polymorphisms revealed that the increased risk can be attributed to the G allele of P1 and/or the 4-bp deletion allele of P3. These alleles are unique to haplotype H-AS3 and so far have not been found in the closely related primates chimpanzee, gorilla, orang-utan, macaque, baboon and wooly monkey, although only one or two animals per species have been studied. These findings suggest that these two alleles occurred late in evolution, probably within a relatively narrow time window on haplotype H-AS1.
Genetic studies by us and others have suggested that the AS-SRO element interacts with the PWS-SRO element to establish the maternal imprint in the female germ line.2, 6, 7, 8 The molecular mechanisms are unknown but likely to involve trans-acting factors that bind to these two elements. It is tempting to speculate that P1 and P3 lie within protein binding sites and that the G and/or the 4-bp deletion allele bind a trans-acting factor with somewhat less efficiency than the A and/or the 4-bp insertion allele. This may increase the risk that the maternal imprint is not or not completely established. If incomplete, the maternal imprint may be lost during the global wave of DNA demethylation during early embryogenesis. In fact, a certain fraction of AS patients with an imprinting defect have somatic mosaicism, and in one case studied the maternal imprint was lost very early.25
Using ES cells and gel-shift experiments, Kantor et al23, 26 have shown protein binding to the AS-SRO and various parts of the PWS-SRO. In the AS-SRO they identified an 18 bp sequence motif (allele discrimination signal, ADS) that was methylated in sperm and unmethylated in oocytes. The differential methylation was maintained in E7.5 old embryos but lost in adults where the ADS is methylated on both alleles. From the methylation changes during development and band shift experiments the authors suggest that this ADS plays a role in the imprinting process and confers imprinting upon the PWS-SRO. The ADS contains the polymorphism P2, which showed no significant P-value in our study. However, the relevance of the protein binding activity in ES cells is unclear. ES cells do not imprint and are unlikely to contain maternal imprinting factors. It would be worthwhile to test the sequences around P1 and P3 for protein binding in human oocytes, but this material is not available in sufficient quantity.
In contrast to the AS-SRO, the PWS-SRO is very little polymorphic. Although it is five times larger than the AS-SRO, which contains five polymorphisms, it contains only one frequent polymorphism 366 bp upstream of exon 1. We did not observe any allele or haplotype in the PWS-SRO region that is associated with an increased risk of an imprinting defect. In contrast to the AS-SRO, the PWS-SRO is not a control element for the establishment of the paternal imprint.27 Furthermore, we have previously found that in PWS patients with a primary imprinting defect the affected chromosome was always inherited from the paternal grandmother.3 This finding suggests that the (grand)maternal imprint was not erased in the paternal germ line. Imprinting erasure may be a passive process or may not require factors binding to the PWS-SRO. Thus, imprint erasure errors may not be affected by sequence variants. Although this explanation is reasonable, we are aware that there are alternative explanations. First, the only frequent SNP within the PWS-SRO (P8) or any rare polymorphism that is in LD with this SNP may in fact be outside the critical element involved in imprint erasure and there is no genetic variation of this element. Second, the sample size may be too small to observe an effect.
We have also obtained tentative evidence that homozygosity for the 677C>T variant of the MTHFR gene might increase the risk of a maternal imprinting defect. The frequency of the 677TT genotype was significantly higher in the mothers of the AS patients than in the patients' fathers or the general population (P=0.028). Assuming the association were genuine, it would have far reaching implications. It would not only suggest that women homozygous for the 677C>T variant are at an increased risk of conceiving a child with an imprinting defect, but that nutrition might also have an affect on imprint establishment or maintenance. Wolff et al28 have shown that folate levels can affect transposon methylation in mice. After folate washout in patients with hyperhomocysteinaemia, Ingrosso et al29 observed biallelic expression of H19, which normally is expressed from the maternal allele. After folate treatment, they observed a shift back to monoallelic expression. It should be noted, however, that the authors did not study the genomic imprints, that is, the methylation patterns directly and that expression changes can occur in the absence of imprint changes.
While it is generally accepted that epigenetic variance adds to genetic variance, there are very few studies on the basis of epigenetic variance. Most of the epigenetic variance is probably stochastic, but as shown by Sandovici et al,9 Murrell et al10 and in this study, some of the epigenetic variance may result from genetic variance in cis-regulatory elements and trans-acting factors. Genetic variation in trans-acting factors such as MTHFR may also interact with environmental cues in modifying the epigenotype.
Accession codes
References
Nicholls RD, Knepper JL : Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2001; 2: 153–175.
Buiting K, Saitoh S, Gross S et al: Inherited microdeletions in the Angelman and Prader–Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet 1995; 9: 395–400.
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B : Epimutations in Prader–Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet 2003; 72: 571–577.
Ohta T, Gray TA, Rogan PK et al: Imprinting-mutation mechanisms in Prader–Willi syndrome. Am J Hum Genet 1999; 64: 397–413.
Bielinska B, Blaydes SM, Buiting K et al: De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat Genet 2000; 25: 74–78.
Dittrich B, Buiting K, Korn B et al: Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat Genet 1996; 14: 163–170.
Shemer R, Hershko AY, Perk J et al: The imprinting box of the Prader–Willi/Angelman syndrome domain. Nat Genet 2000; 26: 440–443.
Perk J, Makedonski K, Lande L, Cedar H, Razin A, Shemer R : The imprinting mechanism of the Prader–Willi/Angelman regional control center. EMBO J 2002; 21: 5807–5814.
Sandovici I, Leppert M, Hawk PR, Suarez A, Linares Y, Sapienza C : Familial aggregation of abnormal methylation of parental alleles at the IGF2/H19 and IGF2R differentially methylated regions. Hum Mol Genet 2003; 12: 1569–1578.
Murrell A, Heeson S, Cooper WN et al: An association between variants in the IGF2 gene and Beckwith–Wiedemann syndrome: interaction between genotype and epigenotype. Hum Mol Genet 2004; 13: 247–255.
Frosst P, Blom HJ, Milos R et al: A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 1995; 10: 111–113.
Friso S, Choi SW, Girelli D et al: A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci USA 2002; 99: 5606–5611.
Stern LL, Mason JB, Selhub J, Choi SW : Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677 T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomarkers Prev 2000; 9: 849–853.
Paz MF, Avila S, Fraga MF et al: Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumors. Cancer Res 2002; 62: 4519–4524.
Castro R, Rivera I, Ravasco P et al: 5,10-methylenetetrahydrofolate reductase (MTHFR) 677C → T and 1298A → C mutations are associated with DNA hypomethylation. J Med Genet 2004; 41: 454–458.
Morgan HD, Santos F, Green K, Dean W, Reik W : Epigenetic reprogramming in mammals. Hum Mol Genet 2005; 14 (Suppl 1): R47–R58.
Zeschnigk M, Lich C, Buiting K, Doerfler W, Horsthemke B : A single-tube PCR test for the diagnosis of Angelman and Prader–Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet 1997; 5: 94–98.
Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Bandelt HJ, Forster P, Rohl A : Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48.
Sham P : Transmission/disequilibrium tests for multiallelic loci. Am J Hum Genet 1997; 61: 774–778.
Spielman RS, Ewens WJ : The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 1996; 59: 983–989.
Buiting K, Dittrich B, Gross S et al: Sporadic imprinting defects in Prader–Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet 1998; 63: 170–180.
Kantor B, Kaufman Y, Makedonski K, Razin A, Shemer R : Establishing the epigenetic status of the Prader-Willi/Angelman imprinting center in the gametes and embryo. Hum Mol Genet 2004; 13: 2767–2779.
van der Put NM, Gabreels F, Stevens EM et al: A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet 1998; 62: 1044–1051.
Nazlican H, Zeschnigk M, Claussen U et al: Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum Mol Genet 2004; 13: 2547–2555.
Kantor B, Makedonski K, Green-Finberg Y, Shemer R, Razin A : Control elements within the PWS/AS imprinting box and their function in the imprinting process. Hum Mol Genet 2004; 13: 751–762.
El-Maarri O, Buiting K, Peery EG et al: Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet 2001; 27: 341–344.
Wolff GL, Kodell RL, Moore SR, Cooney CA : Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J 1998; 12: 949–957.
Ingrosso D, Cimmino A, Perna AF et al: Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003; 361: 1693–1699.
Acknowledgements
We are very grateful to all colleagues and referring clinicians for making DNA samples available to us, Gabriele Gillessen-Kaesbach and Dietmar Lohmann for helpful discussion, Robert Nicholls, Kentaro Yamasaki, Daniel Driscoll and Hui-Jia Dong for communicating unpublished data and Christina Lich for expert technical assistance. Part of this work was supported by the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Zogel, C., Böhringer, S., Groß, S. et al. Identification of cis- and trans-acting factors possibly modifying the risk of epimutations on chromosome 15. Eur J Hum Genet 14, 752–758 (2006). https://doi.org/10.1038/sj.ejhg.5201602
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201602
Keywords
This article is cited by
-
Common genetic variation in the Angelman syndrome imprinting centre affects the imprinting of chromosome 15
European Journal of Human Genetics (2020)
-
Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes
European Journal of Human Genetics (2019)
-
The inheritance of epigenetic defects
medizinische genetik (2017)
-
Genomisches Imprinting und Imprintingfehler
Medizinische Genetik (2010)
-
The C15orf2 gene in the Prader–Willi syndrome region is subject to genomic imprinting and positive selection
neurogenetics (2010)
