
Abstract
We report a novel, heteroplasmic point mutation in the mitochondrial tRNA for tryptophan at position 5532. The mutation was present in all the tissues studied and segregated with the biochemical defect, with higher levels of mutation present in cytochrome c oxidase-deficient muscle fibres. The patient manifested a neurogastrointestinal syndrome with features including failure to thrive, psychomotor retardation, ophthalmoplegia, sensorineural deafness and encephalopathy together with vomiting, diarrhoea and colitis.
Similar content being viewed by others


Introduction
Mitochondrial dysfunction due to defects involving mitochondrial DNA (mtDNA) produces a wide range of clinical phenotypes.1 Classically, these have been associated with disease affecting muscles or other parts of the nervous system, but any and indeed all the tissues may be affected. Symptoms arising from the gastrointestinal tract (G.I.T.) are common in mitochondrial disease, but are often mild and overlooked. The combination of neurological features and serious gastrointestinal disease is uncommon.
More than 80 mitochondrial tRNA mutations have been identified.2 Several occur in what appear to be ‘hotspots’ for mutation such as the tRNA for leucine (UUR); thus far only three mutations have been found in tRNATrp. We describe a young girl with disease affecting both the gastrointestinal and nervous systems in whom investigation identified the presence of a new pathological mtDNA mutation involving tRNATrp.
Case history
The patient, who is now 16 years, was born at term of healthy, nonconsanguineous parents. She presented when aged 1 year with recurrent vomiting and failure to thrive. During the next 2 years she was admitted frequently because of vomiting and poor feeding. From 18 months of age she complained of leg discomfort and was reluctant to run. With minor infections she became drowsy and on one occasion unresponsive. Venous bicarbonate concentrations during this period ranged between 12 and 16 mM (NR 22–28 mM).
From the age of 3 years she regressed cognitively. At 6 years, mobility declined and she became doubly incontinent. Treatment with phenobarbitone was started for suspected epilepsy. Examination at 9 years showed a small, dysmorphic, hirsute girl with very poor dentition; height and weight were below the third percentile. She had sensorineural deafness, ptosis, ophthalmoplegia, pigmentary retinopathy and constricted visual fields. Her muscle bulk was globally reduced, but tone and reflexes were normal. Testing power and coordination were difficult due to poor understanding, but there was a terminal finger intention tremor, slow repetitive hand movements and she fell backwards on standing.
Subsequently, gastrostomy was performed because of feeding difficulties and weight loss. Her hearing worsened and her vision deteriorated to perception of light only. She became mute and immobile and developed spasticity, mainly in her legs. She also developed severe acrocyanosis. From being constipated, she developed diarrhoea with blood and mucous from the age of 13 years. Colonoscopy showed a nonspecific inflammation. Topical steroids were given without benefit, but the symptoms did improve with systemic steroids.
Haemoglobin, blood glucose, bone and liver chemistries were all normal. Lactate concentrations were elevated in blood (3.0 mmol/l; NR <1.7) and cerebrospinal fluid (9 mmol/l; NR <2.0). Renal function remained normal although renal tubular function was never assessed. Echocardiography was normal. Cerebral CT showed generalised atrophy confirmed by MRI, which also showed periventricular white matter changes. Combined skin biopsy and muscle biopsy was performed at the age of 9 years for diagnostic purposes. Consent was given by the parents.
Methods
Muscle histology and histochemistry and biochemical assay of respiratory chain complexes were performed as described previously.3 DNA was extracted from muscle, leukocytes and fibroblasts using standard protocols. Southern blotting, DNA sequencing and last cycle hot and single fibre PCR were performed as previously described.3 Fibroblasts were cultured in the presence of pyruvate and uridine. As the G5532A mutation does not create or destroy a restriction site, we designed mismatch primers that introduce a recognition site for the restriction endonuclease Mbo I. Both primers had an M13-tail (sequence shown in lower case) and recognised the following sequences: Light strand (5360–5379) tgtaaaacgacggccagtCTACTCCACCTCGATCACAC and heavy strand (5552–5533) caggaaacagctatgaccGGGCTTTGAAGGCTCTTGAT (mismatch nucleotides shown in bold). The mismatches present generate two Mbo I restriction sites in the wild-type PCR product. Digestion cuts the 229 bp amplimer into fragments of 160, 39 and 30 bp. The G5532A transition abolishes one Mbo I site, yielding fragments of 199 and 30 bp.
The relative proportion of wild-type and mutant mtDNA genomes determined by the addition of 5 μCi [α-32P]-dCTP (3000 Ci/mmol) prior to the last cycle of the PCR. Labelled products were digested with 10 U Mbo I, separated through a 10% nondenaturing polyacrylamide gel, and the radioactivity in each fragment quantified using the ImageQuant software (Molecular Dynamics).
Results
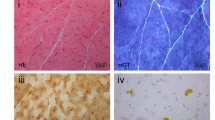
Clinically we suspected a mitochondrial disorder. Muscle biopsy showed 30% ragged-red fibres (RRF) and a mosaic of cytochrome c oxidase (COX) positive and deficient fibres with an extremely high-percentage of deficient fibres (90%). Respiratory chain analysis in isolated muscle mitochondria showed activities less than 20% of control values for both complexes I and IV (data not shown). Southern blotting excluded major mtDNA rearrangement and sequencing of all tRNA genes revealed a heteroplasmic G to A transition at position 5532 (Figure 1a) in the DHU loop of the tRNATrp gene (Figure 1b). This base change is not a recognised polymorphism,2,4 nor has it been seen in the sequence of 65 controls or >50 patients with mitochondrial diseases in whom the genetic defect is unknown. A second, silent change at position 3336 in the ND1 gene was also identified, but no other base change was found.

(a) Sequence analysis of the tRNATrp gene showing the G5532A mutation. (b) The mutation affects the DHU loop potentially allowing further pairing that could lead to steric changes in the tRNA cloverleaf structure. (c) The upper panel shows a cartoon of the RFLP analysis used to assess the percentage of heteroplasmy. The primer sequences are adapted to include Mbo I sites, one of which is lost in the presence of the G5532A mutation. The lower panel shows the results of last-cycle hot PCR analysis in skeletal muscle and COX-positive and negative fibres picked from the biopsy. (d) A graphical presentation of the data derived in panel c showing that the mutation segregates with the biochemical phenotype. COX-negative fibres have significantly higher levels (>90%) of the G5532A mutation (range 91–96%) than COX- positive fibres (range 6–85%; Mann–Whitney U-test, P<0.0001) in which the mutation load varies widely.
Last cycle, hot PCR using the adapted primers (Figure 1c) showed that the mutation was present in all tissues from the patient (muscle, 92%; fibroblast, 37%; blood, 21%) and could also be detected in blood from the patient's mother (7%) and brother (9%) who were clinically unaffected at the time of analysis. Analysis of single fibres picked from sections of muscle sequentially reacted for COX and succinate dehydrogenase showed that the mutation segregated with the biochemical defect with the highest level of mutation in COX-deficient cells (Figure 1c and d).
Discussion
The clinical and muscle biopsy features strongly suggested that our patient had a defect affecting the mitochondrial genome. DNA analysis identified a base substitution at position 5532 in mtDNA in the gene encoding the tRNA for tryptophan. We believe that the G5532A is pathological for the following reasons. The change affects the DHU stem, a functionally important region of the tRNA, at a site that is highly conserved throughout evolution (http://mamit-trna.u-strasbg.fr/). A tRNA defect would, moreover, explain the biochemical findings of multiple respiratory chain complex defects. The change is heteroplasmic, present in maternal relatives and not previously reported. Finally, the G5532A segregates with the phenotype being present at much higher levels in COX deficient fibres than COX positive fibres.
Three other mutations in tRNATrp have been identified. A G to A transition at position 5549 caused adult onset dementia, ataxia, deafness and peripheral neuropathy,5 while a G to A transition at position 5521 caused a late onset myopathy.6 A single T insertion at position 5537 has been found in two families with a Leigh-like phenotype.7,8 Interestingly, the mother of the index child in the first family was also small and thin and had symptoms suggestive of irritable bowel syndrome.
Clinically, our patient had a neurogastrointestinal syndrome. Gastrointestinal symptoms such as vomiting and abdominal discomfort are common in mitochondrial disease,9 but usually overlooked because of serious disease elsewhere. Persistent vomiting and failure to thrive coming on after a disease free interval in infancy, is seen in Leigh's disease, a diagnosis that was considered in our case.
Gastrointestinal dysfunction including recurrent vomiting and pseudo-obstruction has been described with mtDNA mutations, including the A3243G mutation and single mtDNA deletions (reviewed in).9 In one case with encephalopathy and pseudo-obstruction due to bowel dysmotility, the A3243G mutation was found in skeletal muscle and not in the bowel suggesting that the gut dysfunction may have been due to central nervous system disease.10 Enteropathy with villous atrophy is well recognised in patients with mtDNA rearrangements.11 While both eosinophilic and ischaemic colitis have been reported,12,13 colitis remains a very uncommon manifestation in mitochondrial disease.
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is one mitochondrial disorder, in which gastrointestinal features play a central and indeed defining role.14 This rare, autosomal recessive disorder can present from childhood to adulthood with a combination of gastrointestinal (nausea, vomiting, diarrhoea, pain, weight loss and signs of obstruction, liver dysfunction and undernutrition) and neurological features (ptosis, ophthalmoplegia, peripheral neuropathy and leucoencephalopathy).14 While this disorder also produces the hallmarks of mitochondrial disease, namely RRF and COX-deficient fibres, it is caused by a nuclear defect involving the gene for thymidine phosphorylase.15
Our patient demonstrates once again that mitochondrial DNA disease is capable of affecting any and all tissues with the G.I.T being no exception. Clinicians must be alert to this clinical diversity.
References
Chinnery PF, Turnbull DM : Mitochondrial DNA and disease. Lancet 1999; 354 (suppl I): 17–21.
Mitomap: MITOMAP: A Human Mitochondrial Genome Database, http://www.mitomap.org, 2003.
Weber K, Wilson JN, Taylor L et al: A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet 1997; 60: 373–380.
Herrnstadt C, Elson JL, Fahy E et al: Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am J Hum Genet 2002; 70: 1152–1171.
Nelson I, Hanna MG, Alsanjari N, Scaravilli F, Morgan-Hughes JA, Harding AE : A new mitochondrial DNA mutation associated with progressive dementia and chorea: a clinical, pathological, and molecular genetic study. Ann Neurol 1995; 37: 400–403.
Silvestri G, Rana M, DiMuzio A, Uncini A, Tonali P, Servidei S : A late-onset mitochondrial myopathy is associated with a novel mitochondrial DNA (mtDNA) point mutation in the tRNATrp gene. Neuromusc Disord 1998; 8: 291–295.
Santorelli FM, Tanji K, Sano M et al: Maternally inherited encephalopathy associated with a single-base insertion in the mitochondrial tRNATrp gene. Ann Neurol 1997; 42: 256–260.
Tulinius M, Moslemi AR, Darin N et al: Leigh Syndrome with cytochrome-c oxidase deficiency and a single T insertion nt 5537 in the mitochondrial tRNAtrp gene. Neuropediatrics 2003; 34: 87–91.
Bindoff L : Mitochondrial dysfunction and the gastrointestinal system; in Desneulle C, DiMauro S (eds): Mitochondrial disorders: from pathophysiology to acquired disorders, Berlin: Springer; 2002, pp 275–285.
Chinnery PF, Jones S, Sviland L et al: Mitochondrial enteropathy: the primary pathology may not be within the gastrointestinal tract. Gut 2001; 48: 121–124.
Cormier-Daire V, Bonnefont J-P, Rustin P et al: Mitochondrial DNA rearrangements with onset as chronic diarrhea with villous atrophy. J Pediatr 1994; 124: 63–70.
Morris AA, Lamont PJ, Clayton PT : Pearson's syndrome without marrow involvement. Arch Dis Child 1997; 77: 56–57.
Hess J, Burkhard P, Morris M, Lalioti M, Myers P, Hadengue A : Ischaemic colitis due to mitochondrial cytopathy. Lancet 1995; 346: 189–190.
Hirano M, Silvestri G, Blake DM et al: Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Neurology 1994; 44: 721–727.
Nishino I, Spinazzola A, Hirano M : Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999; 283: 689–692.
Acknowledgements
RWT, ZC-L and DMT acknowledge the continuing support of the Muscular Dystrophy Campaign and The Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maniura-Weber, K., Taylor, R., Johnson, M. et al. A novel point mutation in the mitochondrial tRNATrp gene produces a neurogastrointestinal syndrome. Eur J Hum Genet 12, 509–512 (2004). https://doi.org/10.1038/sj.ejhg.5201185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201185
Keywords
This article is cited by
-
Molecular pathomechanisms and cell-type-specific disease phenotypes of MELAS caused by mutant mitochondrial tRNATrp
Acta Neuropathologica Communications (2015)
-
The tRNAGly T10003C mutation in mitochondrial haplogroup M11b in a Chinese family with diabetes decreases the steady-state level of tRNAGly, increases aberrant reactive oxygen species production, and reduces mitochondrial membrane potential
Molecular and Cellular Biochemistry (2015)
-
Mitochondrial myopathy associated with a novel 5522G>A mutation in the mitochondrial tRNATrp gene
European Journal of Human Genetics (2013)
-
Gastrointestinal and hepatic manifestations of mitochondrial disorders
Journal of Inherited Metabolic Disease (2013)
-
Functional consequences of mitochondrial tRNATrp and tRNAArg mutations causing combined OXPHOS defects
European Journal of Human Genetics (2010)
