
Abstract
Prader–Willi syndrome (PWS) and Angelman syndrome (AS) are associated with a loss of function of imprinted genes in the 15q11–q13 region mostly due to deletions or uniparental disomies (UPD). These anomalies usually occur de novo with a very low recurrence risk. However, in rare cases, familial translocations are observed, giving rise to a high recurrence risk. We report on the difficulties of genetic counseling and prenatal diagnosis in a family segregating for a translocation (14;15)(q11;q13) where two consanguineous parents carry the same familial translocation in this chromosome 15 imprinting region. Both children of the couple inherited a chromosomal anomaly leading to PWS. However, a paternal 15q11–q13 deletion was responsible for PWS in the first child, whereas prenatal diagnosis demonstrated that PWS was associated with a maternal 15q11–q13 UPD in the fetus. This report demonstrates that both conventional and molecular cytogenetic parental analyses have to be performed when a deletion is responsible for PWS or AS in order not to overlook a familial translocation and to insure reliable diagnosis and genetic counseling.
Similar content being viewed by others
Introduction
The Prader–Willi syndrome and the Angelman syndrome are two clinically distinct syndromes that are associated with a loss of function of imprinted genes in the same chromosomal region. Most of the patients with PWS or AS (70–75%) have a cytogenetic deletion of chromosome 15q11–q13 region that is paternal in PWS and maternal in AS.1,2,3,4 Chromosome 15 maternal uniparental disomy (UPD) is responsible for almost all the remaining PWS patients;5 by contrast, paternal UPD is responsible for only 1–5% AS patients, whereas 20–30% result from either imprinting defects or point mutations or small deletions within the maternal UBE3A gene.6 Most of deletions observed in PWS or AS are de novo interstitial deletions probably due to unequal crossover between repeated DNA sequences;7 however, they can sometimes (5%) result from structural rearrangements involving the 15q11–q13 region such as duplications, small bisatellited additional chromosomes, inversions and/or translocations.8 These anomalies usually occur de novo; however, in rare cases, they may be the result of an unbalanced cryptic structural chromosome rearrangement inherited from a parental inversion9 or by segregation of chromosomes involved in familial translocations. To our knowledge, only three families have been reported in which both PWS and AS were observed in close relatives due to the malsegregation of a balanced familial translocation involving chromosome 15: in one family,10 the index child showed AS, whereas two previously reported relatives11 had PWS. In another case,12 two cousins were initially diagnosed with PWS; nevertheless, as one patient displayed seizures and severe developmental delay and inherited the abnormal chromosome 15 from his mother, he probably had an AS, as suggested later on.13 In a third family with a familial balanced translocation between chromosomes 6 and 15,14 one of two first cousins displayed PWS and the other had AS; PWS resulted from a deletion in the paternally derived chromosome 15 and AS was due to chromosome 15 paternal UPD.
We report here the difficulties of genetic counseling and prenatal diagnosis in a new family including close relatives with PWS and AS due to a familial balanced translocation t(14;15)(q11;q13).
Case report
A young and healthy Caucasian couple (IV4–IV13) was referred for genetic counseling and prenatal diagnosis because their first child (V1) was affected with typical PWS including hypotony and poor sucking reflex at birth, secondary hyperphagia with obesity, small hands and feet, micropenis, learning disability and moderate mental retardation. A reciprocal translocation between chromosome 14 and chromosome 15 had been previously identified in the father's family (Figure 1) because of the presence of two malformative syndromes. Patient V14 showed classic symptoms of AS with microcephaly, severe mental retardation, ataxic gait with jerky arm movements, seizures, electroencephalogram abnormalities and paroxysms of laughter; patient V11 had a complex malformation syndrome including macrocephaly and ventricular dilatation, frontal bossing, downslanting eyes, micrognathism, large ears, hypotony and mental retardation. The diagnosis of AS was subsequently ascertained in V3 who displayed seizures and severe developmental delay. Both children with AS had a 15pter → 15q13 deletion as well as the PWS patient. Patient V11 had a 15pter → 15q13 trisomy. Individuals III6, IV6, IV11, IV13, IV17, V4, V9, V10 and V13 were phenotypically normal carriers of the balanced translocation. As the pregnant woman (IV4) and her husband (IV13) were consanguineous, she was at risk of carrying the familial translocation. Chromosomal analysis was performed and showed that IV4 had the same reciprocal translocation as her husband. The overall risk estimation was evaluated taking into account both the risk of malsegregation of the two parental translocations and the risk due to 15q11–q13 genomic imprinting (Table 1). After informing the parents, prenatal diagnosis was carried out first on chorionic villi by cytogenetic and indirect molecular approaches, and secondly on fetal blood because methylation analysis was necessary. As PWS was identified in the fetus, the parents opted for termination of pregnancy.

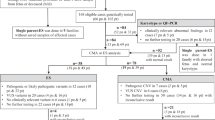
Pedigree showing segregation of the balanced translocation t(14;15)(q11;q13) and microsatellite genotypes in the AS/PWS region. The three chromosome 15 microsatellite markers are arranged from centromeric (top) to telomeric (bottom): IR4-3R (D15S11), LS6-1 (D15S113)1 and GABRB3. (LS6-1 microsatellite has to be interpreted with caution because of possible nonamplified alleles; however, it was fully informative in the reported family).
Cytogenetic studies
Parents
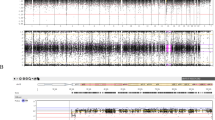
High-resolution chromosomes at the 550 band stage were obtained from peripheral blood lymphocytes using a fluorodeoxyuridine (FdU) synchronization technique followed by the releasing of the block in DNA synthesis with thymidine. Chromosomes were G-banded with trypsin (GTG technique) and R-banded by incubation in hot saline solution followed by Giemsa staining (RHG technique). The chromosomes of the pregnant woman (IV4) revealed the same translocation as those of her husband (IV13) and could be defined as 46,XX, t(14;15)(q11;q13) (Figure 2).

Partial karyotype of the IV4 translocated mother with 14 and 15 ideograms
Fluorescent in situ hybridization studies on metaphase spreads from peripheral blood lymphocytes confirmed conventional cytogenetic analysis. These analyses were only recently performed as they were not available at the time of this evaluation. Probes for loci SNRPN and D15S10 from Vysis (both are associated with D15Z1 probe) and SNRPN from Oncor were used and protocols recommended by the manufactures were applied. All three probes showed hybridization signals on both normal 15 chromosome and der (15) chromosome. The distal probes (15qter control probe from Oncor and PML from Vysis) appeared on a different D-group chromosome.
Prenatal diagnosis
At 12 weeks' gestation, a first conventional chromosomal analysis was performed after chorionic villus sampling by cytotrophoblast direct analysis and long-term culture technique set up by chorionic villi enzymatic dissociation. Chromosomes were R-banded. Direct chromosome analysis (20 mitoses) showed a ‘46,XY’ karyotype, and R-banding cytogenetic analysis on cultured villi (20 mitoses) was very suggestive of a balanced karyotype 46,XY, t(14;15)(q11;q13).
At 24 weeks' gestation, high-resolution R-banded chromosomes obtained from fetal blood with the same technique used for the parents confirmed the balanced translocation in the fetus.
Molecular studies
Familial analysis
DNA was isolated from peripheral blood leukocytes of AS (V3, V14) and PWS (V1) patients and of all members of the family at risk of the balanced translocation (Figure 1). Indirect analysis with three microsatellite markers distal to the translocation breakpoint IR4-3R (D15S11), LS6-1 (D15S113) and GABRB315 confirmed the cytogenetic results (Figure 1).
Prenatal diagnosis
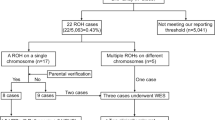
DNA was isolated from chorionic villi and indirect analysis was performed with the three familial informative markers. The fetal parental-origin specific DNA methylation imprint was determined by a HindIII+HpaII Southern blot hybridized with the PW71B probe16 at the D15S63 locus, which was the only available methylation test at the time of this evaluation. Nevertheless, as the PW71B probe reliability was not established in chorionic villi as well as in amniocytes due to possible tissue hypomethylation in early fetal samples, methylation analysis was also performed on fetal blood. Indeed, analysis of chorionic villi DNA was inconclusive, while a pattern characteristic of PWS was observed in fetal blood DNA (Figure 3). Different reports subsequently demonstrated that the PW71B probe should not be used for prenatal diagnosis because of possible inconsistent methylation pattern at this D15S63 locus, whereas the methylation status of the exon alpha of the small nuclear ribonucleoprotein-associated polypeptide N (SNRPN) gene is stable (KB17 probe).17,18 However, our result was certain as it was confirmed on fetal tissues after abortion both with the PW71B probe and, later on, with the KB17 probe.

Methylation analysis of the D15S63 locus with the PW71B probe. The parental origin of the HindIII+HpaII bands are indicated: maternal (mat) and paternal (pat). (A) PWS patient with a paternal deletion; (B) normal control; (C) fetus DNA isolated from fetal blood; (D) fetus DNA isolated from chorionic villi. A pattern characteristic of PWS is detected on DNA isolated from fetal blood, whereas analysis on chorionic villi was inconclusive due to hypomethylation.
Discussion
In the family described in this report, PWS and AS result from a familial cryptic translocation between chromosome 15 (q13 region) and chromosome 14 (q11 region). In several other PWS or AS reports, deletions also result from a cryptic translocation between 15q11–q13 AS–PWS region and another proximal q11–q12 acrocentric chromosome region in one of the parents: 22q11,10,11,19 13q12.320 and 14q11.2.12,21 As previously reported, a 15q11–q13 cytogenetic abnormality is difficult to reliably detect due to the small size of the region, its proximity to the centromere and the existence of 15q11.2 region heteromorphism.8 In the reported case, a familial translocation was suspected because patient V11 had a distinct phenotype from the patient with AS (V14), indicating the presence of both unbalanced rearrangements. A cryptic translocation between chromosome 14 and chromosome 15 could be recognized on high-resolution R-banding karyotyping because the translocated regions were differently dark R-stained (Figure 2). In the other reported cases with cryptic acrocentric translocations, the discovery of the translocation failed with high-resolution G-banding: for the oldest cases,11,12 only Distamycin/DAPI fluorescence banding, which is characteristic of chromosome 15 short arm, ascertained the diagnosis; for the more recent reports,19,20,21 only fluorescent in situ hybridization (FISH) analysis using a Prader–Willi Angelman probe associated with an alpha-satellite probe and distal control probes for chromosome 15 demonstrated the cryptic translocation. As it is of most importance to identify a cryptic translocation for genetic counseling and reliable prenatal diagnosis, these authors insist on the necessity to systematically use an alpha-satellite centromere probe for chromosome 15 or a probe proximal to the 15q11–q13 critical region in combination with the 15q11–q13 and distal control probes.
In the reported family, as the parents were consanguineous and carried the same familial translocation, the theoretical risks of offspring have to be evaluated taking into account both the risk of an unbalanced genome due to 15q11–q13 region malsegregation and the risk due to 15q11–q13 genomic imprinting (Table 1). The lack of phenotypic effects of the malsegregation of the 14pter → q11 region, which includes chromosome 14 satellites, nucleolar organiser and centromere, may be reasonably overlooked as this region is mainly heterochromatic as previously suggested for other acrocentric chromosomes.10 Of the possible parental meiotic segregation modes, 3:1 malsegregations could theoretically occur but are unlikely as they require two events with either a trisomy rescue to correct a trisomy 14 or 15 or a segmental gamete complementation to correct a monosomy 14 or 15. The latter would result in UPD 15q13-qter without phenotypical consequences or in UPD 14q11-qter where a phenotype has to be considered. As they require only one event, the most likely segregation modes are 2:2 segregations with either alternate 2:2 segregation resulting in balanced gametes or adjacent segregation leading to chromosomal inbalances. Adjacent-2 malsegregation – where adjacent homologous centromeres go to the same cell daughter – is more likely to occur as pairing between homologous regions has to be maximized and both translocated centric segments are small in content.22,23,24 In fact, it was the unique mode of malsegregation observed within the reported pedigree (V1, V3, V11, V14). The overall risk of offspring is summarized in Table 1. Of the 16 theoretical possibilities, only four (25%) with alternate segregation result in normal offspring: one with normal karyotype (Table 1: 1), two with one of the balanced parental translocations and no UPD (Table 1: 4,13) and one with the two balanced parental translocations (Table 1: 16). Given the high risk of abnormal offspring, chorionic villi sampling was proposed to the parents as ten defavorable issues could be eliminated either with conventional cytogenetic or microsatellite molecular studies (Table 1: 2, 3, 5, 6, 8, 9, 11, 12, 14, 15). Based on segregation analysis of GABRB3 alleles, the fetal haplotype was compatible with three of the 16 possibilities: two balanced translocations (Table 1: 7, 13) and a 15pter → 15q13 trisomy (Table 1: 15). As chromosome analysis on chorionic villi cultures demonstrated a 46 XY, t(14;15)(q11;q13) karyotype, 15pter → 15q13 trisomy (Table 1: 15) could be excluded by cytogenetics. Of the four situations associated with only one of the parental balanced translocations (Table 1: 4, 7, 10, 13), two (Table 1: 4, 10) could be excluded by microsatellite analysis. Finally, after chorionic villi cytogenetic and molecular analyses, the two remaining possibilities were either a balanced translocation with a normal phenotype (Table 1: 13) or a balanced translocation with maternal UPD for chromosome 15 corresponding to PWS (Table 1: 7). As distinctive methylation patterns are observed in normal and PWS individuals, methylation analysis had to be explored in order to distinguish between a balanced translocated fetus with a normal phenotype and a PWS fetus. As chorionic villi were unsuitable for methylation analysis, fetal blood sampling was performed. Unfortunately, the methylation profile revealed loss of paternal methylation pattern at locus D15S63 and a pattern characteristic of only the maternal chromosome 15, corresponding to a PWS fetus due to maternal UPD. Finally, both children of the couple IV4–IV13 inherited a chromosomal anomaly leading to PWS. However, PWS occurred by two different mechanisms: in patient V1, PWS was associated with a paternal 15q11–q13 deletion (Table 1: 5) corresponding to adjacent-2 malsegregation, whereas fetus V2 carried a maternal 15q11–q13 UPD (Table 1: 7).
This report demonstrates once more the clinical consequences of 15q11–q13 genomic imprinting and its importance for reliable genetic counseling. It provides a new example that PWS or AS may result from balanced cryptic translocations involving 15q11–q13 region and, frequently, a proximal long-arm region of another acrocentric chromosome. Given the difficulties to recognize these translocations with conventional cytogenetics, it is imperative, in presence of PWS or AS, to systematically control parental chromosome 15 structure with molecular cytogenetic method.
This unique case with consanguineous parents carrying the same reciprocal translocation in an imprinting region emphasizes the importance of an accurate genetic counseling given the risk of 75% of unfavourable outcomes either due to the malsegregation of parental translocations or to genomic imprinting. It also underlines the difficulties of prenatal diagnosis that must associate precocity and reliability: in the present case, even if fetal blood analysis had been performed, chorionic villi sampling was justified as 14 of the 16 possible outcomes could have been eliminated after chorionic villi studies.
Lastly, this report shows the necessity of the association of conventional and molecular cytogenetic analyses and molecular studies to insure reliable prenatal diagnosis.
References
Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD : Deletions of chromosome 15 as a cause of the Prader Willi syndrome. N Engl J Med 1981; 304: 325–359.
Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M : Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader Willi syndrome. Nature 1989; 342: 281–285.
Magenis RE, Brown MG, Lacy DA, Budden S, LaFranchi S : Is Angelman syndrome an alternate result of del(15)(q11–q13)? Am J Med Genet 1987; 28: 829–838.
Malcolm S, Clayton-Smith J, Nichols M et al: Uniparental disomy in Angelman's syndrome. Lancet 1991; 337: 694–697.
Nicholls RD : New insights reveal complex mechanisms involved in genomic imprinting. Am J Hum Genet 1994; 54: 733–740.
Engel E, Antonorakis SE (eds) The Angelman syndrome; in: Genomic imprinting and uniparental disomy in medicine. Clinical and molecular aspects. New York: Wiley-Liss; 2002, pp 187–209.
Horsthemke B, Maat-Kievit A, Sleegers E et al: Familial translocations involving 15q11–q13 can give rise to interstitial deletions causing Prader–Willi or Angelman syndrome. J Med Genet 1996; 33: 848–851.
Knoll JH, Wagstaff J, Lalande M : Cytogenetic and molecular studies in the Prader–Willi and Angelman syndromes: an overwiew. Am J Med Genet 1993; 46: 2–6.
Clayton-Smith J, Driscoll DJ, Waters MF et al: Difference in methylation patterns within the D15S9 region of chromosome 15q11–13 in first cousins with Angelman syndrome and Prader–Willi syndrome. Am J Med Genet 1993; 47: 683–686.
Hulten M, Armstrong S, Challinor P et al: Genomic imprinting in an Angelman and Prader–Willi translocation family. Lancet 1991; 338: 638–639.
Fernandez F, Berry C, Mutton D : Prader–Willi syndrome in siblings, due to unbalanced translocation between chromosomes 15 and 22. Arch Dis Child 1987; 62: 841–843.
Hasegawa T, Hara M, Ando M et al: Cytogenetic studies of familial Prader–Willi syndrome. Hum Genet 1984; 65: 325–330.
Schinzel A, Robinson WP, Bottani A, Yagang X, Prader A : Prader–Willi or Angelman syndrome in familial 15q11 → q13 deletion of maternal origin? Hum Genet 1992; 88: 361–362.
Smeets DFCM, Hamel BCJ, Nelen MR et al: Prader–Willi syndrome and Angelman syndrome in cousins from a family with a translocation between chromosomes 6 and 15. N Engl J Med 1992; 326: 807–811.
Mutirangura A, Greenberg F, Butler MG et al: Multiplex PCR of three dinucleotide repeats in the Prader–Willi/Angelman critical region (15q11–q13): molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet 1993; 2: 143–151.
Dittrich B, Robinson WP, Knoblauch H et al: Molecular diagnosis of the Prader–Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11–q13. Hum Genet 1992; 90: 313–315.
Kubota T, Aradhya S, Macha M : Analysis of parent of origin specific DNA methylation at SNPRN and PW71 in tissues: implication for prenatal diagnosis. J Med Genet 1996; 33: 1011–1014.
Glenn CC, Saitoh S, Jong MT et al: Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet 1996; 58: 335–346.
Missirian C, Malzac P, Arfi J et al: Cryptic translocation resulting in Angelman syndrome: implication for genetic counselling (abstract). Eur J Hum Genet 2002; 10 (Suppl 1): 227.
Tepperberg JH, Tennison MB, Kaiser-Rogers K, Albright SG, Aylsworth AS, Rao KW : An inherited cryptic translocation between chromosomes 13 and 15, detected by FISH in a child with Angelman syndrome (abstract). Am J Hum Genet 1993; 53 (Suppl): 609.
Burke LW, Wiley JE, Glenn CC et al: Familial cryptic translocation resulting in Angelman syndrome: implications for imprinting or location of the Angelman gene? Am J Hum Genet 1996; 58: 777–784.
Jalbert P, Sele B : Factors predisposing to adjacent 2 and 3: 1 disjunctions: study of 161 human reciprocal translocations. J Med Genet 1979; 16: 467–478.
Jalbert P, Sele B, Jalbert H : Reciprocal translocations: a way to predict the mode of imbalanced segregation by pachyten-diagram drawing. A study of 151 human translocations. Hum Genet 1980; 55: 209–222.
Duckett DP, Roberts SH : Adjacent 2 meiotic disjunction. Report of a case resulting from a familial 13q;15q balanced reciprocal translocation and review of the literature. Hum Genet 1981; 58: 377–386.
Acknowledgements
We thank Sylvie Friedmann, Mireille Nuss and Evelyne Schmitt for their technical assistance. This work was supported by the Centre Régional d'Alsace et de Moselle d'Etudes Prénatales (CRAMEP).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flori, E., Biancalana, V., Girard-Lemaire, F. et al. Difficulties of genetic counseling and prenatal diagnosis in a consanguineous couple segregating for the same translocation (14;15) (q11;q13) and at risk for Prader–Willi and Angelman syndromes. Eur J Hum Genet 12, 181–186 (2004). https://doi.org/10.1038/sj.ejhg.5201134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201134
