
Abstract
We report on a patient with severe growth retardation and IgF1 deficiency, in which a mitochondrial abnormality was suspected. An isolated mitochondrial respiratory chain complex III deficiency was found in blood lymphocytes and skin fibroblasts. Sequence analysis of the cytochrome b, which is the only mitochondrial DNA-encoded subunit of complex III, revealed a homoplasmic G15498A mutation, resulting in the substitution of a highly conserved amino acid (glycine 251 into an aspartic acid). The mutation was found to be homoplasmic in all tissues examined from the mother and her brother (lymphocytes, fibroblasts, hair roots and buccal cells). Complex III deficiency was also demonstrated in these cells. Nevertheless, the mother and the brother were asymptomatic. This mutation had been considered as a cardiomyopathy-generating mutation in a previously reported case, and its pathogenicity has been demonstrated recently in yeast. However, it seems not to fulfil the classical criteria for pathogenicity of a mitochondrial DNA mutation, especially the heteroplasmic status, and to be clinically silent, albeit present, in nonaffected relatives. We suggest that other factors are contributing to the clinical variability expression of the G15498A mtDNA mutation.
Similar content being viewed by others


Introduction
Mitochondrial respiratory chain defects are a genetically heterogeneous group of diseases with a wide spectrum of clinical manifestations.1 Ubiquinol-cytochrome c oxidoreductase (EC 1.10.2.2) or complex III (CIII) deficiency represents a relatively rare cause of respiratory chain dysfunction.2 CIII catalyses electron transfer between coenzyme Q and cytochrome c, thereby translocating protons across the mitochondrial inner membrane. CIII is composed of 11 subunits, among them cytochrome b (cyt b) is the only mitochondrial DNA (mtDNA)-encoded subunit. The cyt b gene is located at nucleotide position 14 747–15 887 of the mtDNA, and is flanked by the transfer RNA genes for glutamic acid and threonine. It encodes a protein of 380 amino acids (AA).
Mutations in the cyt b gene with a decrease of respiratory chain CIII activity have been frequently described in patients with cardiomyopathy3,4 or exercise intolerance.5,6,7 Recently, mutations in the nuclear genes encoding the assembling complex III protein8,9 or the structural QP-C subunit of complex III10 have been described.
The metabolic work-up of a patient with severe growth retardation led us to uncover an isolated CIII deficiency in his lymphocytes and fibroblasts. Our purpose was to screen for a mutation of the cyt b gene in this patient and to look for its maternal inheritance, in order to have a molecular evidence of the respiratory chain dysfunction.
Material and methods
Case report
The patient, a 3-year-old boy, was born to nonconsanguineous parents after a term pregnancy and normal delivery. Except for a syndactily of toes and a left dorsal scoliosis, his development was normal until the age of 6 months. He then presented with progressive failure to thrive, and insufficient weight gain (both <5° percentile), despite a correct alimentation. Gastroenterologic work-up showed no malabsorption. His psychomotor development was normal. At 11 months of age, routine investigations uncovered a mild alteration of biological liver tests, which resolved spontaneously. A low serum IgF1 level without growth hormone (GH) deficiency was demonstrated. A mild form of Smith–Lemli–Opitz syndrome was excluded by serum 7-dehydrocholesterol assay. Blood levels of lactate, pyruvate, ketone bodies and lactate CSF and lactate/pyruvate molar ratios were normal. Plasma and urine chromatography of amino acids and urine chromatography of organic acids did not reveal any abnormalities. At 8 months of age, cerebral magnetic resonance imaging was difficult to interpret, but was considered as normal at 18 months of age.
Methods
The study has been approved by the local ethics committees and parents have given their written informed consent.
Skin fibroblasts of the patient and of his mother were grown in HAM'S F10 medium supplemented with 10% foetal calf serum and 200 μ M uridine. Oxygen consumption was measured with a Clark oxygen electrode (Hansatech, UK) in a 250 μl cell, thermostated at 37°C, according to Rustin et al.11
Enzyme activities
Cytochrome c oxidase (CIV: EC 1.9.3.1), antimycin-sensitive decylubiquinol cytochrome c reductase (CIII: EC 1.10.2.2), succinate ubiquinone dichlorophenolindophenol (DCIP) reductase (CII: EC 1.3.99.1) activities were measured spectrophotometrically in lymphocytes and fibroblasts, according to Rustin et al.11
Results were expressed both as specific activities and as ratios between activities.
Sequence analysis of the cyt b gene
Total DNA extracted from lymphocytes or from cultured skin fibroblasts (according to conventional methods) was submitted to polymerase chain reaction (PCR) amplification, by using four pairs of specific primers designed from the genomic sequence of the mitochondrial cyt b gene (Cambridge reference sequence12).
Forward primers and reverse primers (5′ → 3′) were as follows: pair 1 (14 690–14 709 and 15 024–15 005), pair 2 (14 951–14 970 and 15 339–15 320), pair 3 (15 269–15 288 and 15 610–15 591) and pair 4 (15 554–15 573 and 15 934–15 915).
PCR amplification was performed in a 50 μl volume containing 100 ng DNA, 10 pmol of each primer, 25 mM MgCl2, 0.2 mM dNTP and 0.5 U Taq polymerase. The amplification conditions included 3 min at 94°C, 30 cycles of 30 s at 94°C, 60 s at 52°C, 1 min 30 s at 72°C, 10 min at 72°C and hold at 10°C.
For sequence analysis, amplification products were purified with the high Pure PCR Product purification kit (Boehringer Mannheim). Direct sequencing of the PCR products was performed by using 1 pmol amplification primer, 100 ng DNA, 4 μl sequencing reaction mixture (Big Dye Terminator, Roche) and analysis on an automated ABI PRISM 310R sequence analyzer.
PCR and restriction fragment length polymorphism (RFLP) analysis
Frequent mtDNA mutations (MELAS 3243 and 3271, MERRF 8344 and 8356) were screened by PCR and RFLP and large rearrangements were screened by long-range PCR (Boehringer Manheim).
Since the G to A transition at 15 498 nt eliminates a restriction site for StyI, this enzyme was used in RFLP analysis (PCR with pair 3 of primers). PCR amplification products from buccal cells or hairs roots (three) were obtained after an overnight treatment with proteinase K in 100 μl TRIS-EDTA buffer containing 0.05% Tween 20. The products were submitted to electrophoresis through a 2% agarose gel for 1 h 30 s, and revealed using ethidium bromide.
Results
Enzyme analyses
Respiratory chain activities were measured in lymphocytes and fibroblasts of the patient and his healthy mother, and in lymphocytes of his healthy brother (Table 1). Results were expressed both as specific activities and as ratio between activities. Complex III activities were decreased or at the lowest value of the control range. In all cases, activity ratios involving complex III, which best reflects the balanced respiratory chain enzyme activity, were also abnormal. Accordingly, low rates of oxygen consumption in permeabilized cells were demonstrated for decyl-ubiquinol by polarographic measurement. Complex IV values were apparently low in fibroblasts, but might be considered as normal when expressed as a ratio to citrate synthase activity. These results indicated that an isolated complex III deficiency was present in cells of our patient, his brother and mother. To note, a slight increase of citrate synthase activity was observed in the asymptomatic mother and brother, which might denote some type of compensatory mechanism.
Molecular analyses
Sequence analysis of the cyt b gene of the patient and alignment of his nucleotide sequence with the Cambridge reference sequence12 led us to identify common polymorphisms13: A14793G, changing a histidine into an arginine; A15218G, changing a threonine into an alanine; A15326G, changing a threonine into an alanine. Another mutation was also found: a G15498A transition, changing a glycine (251) into an aspartic acid. The same polymorphisms and the G15498 mutation were found in the lymphocytes and fibroblasts of the mother and in the lymphocytes of the brother.
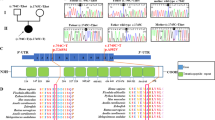
StyI restriction analysis showed that the 15498 mutation was homoplasmic in both lymphocytes and fibroblasts from the patient and his mother and in lymphocytes of his brother (Figure 1a). Screening for heteroplasmy was negative in hair roots and buccal cells from the patient, her mother and her brother (not shown) The 15498 mutation was not detected from over 120 unrelated controls.

Restriction analysis showing the G15498A transition (a) and evolutionary comparison of the cyt b amino-acid sequence around AA251 (b); PCR products were analysed in 2% agarose gel and revealed with ethidium bromide. 1: PCR-amplified fragments with pair 3 of primers before digestion with StyI; 2, 3, 4, 5, 6: PCR amplified fragments after digestion with StyI; 1, 3: lymphocytes of the patient; 2: lymphocytes of a control; 4: fibroblasts of the patient; 5: lymphocytes of the patient's mother; 6: lymphocytes of the patient's brother. When the mutation is present, StyI digestion of the 341 bp fragment generates three smaller fragments of 123, 117 and 101 bp. StyI digestion of the 341 bp fragment generates only two smaller fragments of 240 and 101 bp with control DNA.
Discussion
Complex III deficiency was found in lymphocytes and fibroblasts of a boy with severe growth retardation. Previously, complex III deficiency has been demonstrated in cases of cardiomyopathy,3,4 exercise intolerance5,6,7,14 or encephalomyopathy.15 In these cases, the defect was essentially expressed in one tissue, and was shown to result from anomalies of mtDNA. The possibility of an mtDNA alteration in our patient was investigated by looking for frequent tRNA mutations, large-scale mtDNA rearrangements and Cyt b mutations. A homoplasmic mutation of Cyt b gene at 15 498 nt was found in all examined tissues, including hair roots.
This mutation fulfils some criteria for pathogenicity: it affects a very highly conserved amino-acid stretch16 (Figure 1b). It converts a neutral glycine to an acidic aspartic acid, which is located close to the ubiquinone-binding site, needed for proton transfer to occur. It has never been reported as a polymorphism and was not detected in over than 120 controls.
Two previous cases of complex III deficiency with mutations in the same region of mtDNA have been previously reported. A 24 bp deletion (from 15 498 to 15 521) resulting in the loss of 251 to 258 aa has been shown to be linked to the loss of complex III activity in a patient with exercise intolerance.17 The A15498G mutation was described to be the cause of a revisited case of histiocytoid cardiomyopathy, in a patient whose complex III deficiency has been demonstrated a long time ago in a muscle heart biopsy.18,19 The mutation was found heteroplasmic in this case. Recently, pathogenicity of the A15498G mutation has been demonstrated in yeast.20 The authors have found that this mutation leads to a thermosensitive complex III activity. It disturbs the respiratory function of cells grown at high temperature (36°C), and renders the noncatalytic subunit Qcr9p essential for the activity of bc1 complex at 28°C.
Paradoxically, our patient has severe growth retardation, but his brother and his mother are asymptomatic, despite the fact that they also present a complex III defect associated to a homoplasmic level of the G15498A mutation in all tissues examined. Noticeably, the sequencing of the Cyt b in the mother and the brother did not reveal any other mutation. These results suggest that compensatory mechanisms could interfere to determine the clinical phenotype associated with complex III deficiency. Compensatory mechanisms might involve slight changes in mitochondrial activity, as those possibly denoted by citrate synthase increased. On the other hand, besides decreased ATP production, the consequence of Cyt b mutation may be an overproduction of superoxides, whose generation and fate presumably depend on both environmental and genetic factors, different between individuals. In keeping with this, nuclear encoded compounds might act as modifying factors in such condition. Accordingly, nuclear factors have been proposed to control the different clinical presentations of patients with homoplasmic mutations associated to cases of Leber optic neuropathy (LHON),21 maternally inherited type II diabetes22 and the A1555G mutation of the mitochondrial 12S ribosomal RNA gene.23 Similarly, the A15498G Cyt b gene mutation could be another homoplasmic mutation whose phenotypic expression might be dependent on the interaction of several genes. Another unidentified mutation in a paternally inherited nuclear modifying factor could have had modulatory effects on the phenotypic expression of the disease in this family. In keeping with this, it have been shown in the yeast Saccharomyces cerevisiae that the association between the A15498G mutation and the deletion of the nuclear Qcr9p gene is needed to observe a growth defect at 28°C.20
Growth retardation is frequently associated with respiratory chain dysfunction and IGF1 deficiency might be the consequence of the metabolic disturbance in the affected boy, but the puzzling data are the absence of severe clinical problems in his mother and brother. It could be hypothesized that compensatory pathways balance the complex III defect in these individuals. Such hypothesis has been recently suggested to explain the difference between the clinical phenotypes in a family with a homoplasmic mtDNA mutation and low level of complex IV activity.24 The mother and her children had an homoplasmic level of mtDNA mutation and complex IV deficiency in fibroblasts and muscle. However, all children died in the neonatal period or infancy from a progressive neurological deterioration, whereas the mother seemed healthy and has only one surviving child from 10 pregnancies.
Our work emphasizes the difficulty to establish phenotype/genotype correlations in patients with mtDNA mutations. The same A15498G mutation of Cyt b has led to a severe form of cardiomyopathy in a previously reported patient, and to a mild clinical phenotype with either growth retardation or complete lack of clinical signs in our observation.
Abbreviations
- MELAS:
-
mitochondrial myopathy encephalopathy Lactic Acidosis Stroke-like episode
- MERRF:
-
Myoclonic Epilepsy Ragged Red Fiber
References
Mourmans J, Wendel U, Bentlage HACM et al: Clinical heterogeneity in respiratory chain complex III deficiency in childhood. Neurol Sci 1997; 149: 111–117.
Valnot I, Kassis J, Chretien D et al: A mitochondrial cytochrome b mutation but no mutations of nuclearly encoded subunits in ubiquinol cytochrome c reductase (complex III) deficiency. Hum Genet 1999; 104: 460–466.
Marin-Garcia J, Ananthakrishnan R, Gonzalvo A, Goldenthal MJ : 1995 Novel mutations in mitochondrial cytochrome b in fatal post partum cardiomyopathy. J Inher Metab Dis 1995; 18: 77–78.
Marin-Garcia J, Hu Y, Ananthakrishnan R, Perpont ME, Pierpont GL, Goldenthal MJ : A point mutation in the cyt b gene of cardiac mtDNA associated with complex III deficiency in ischemic cardiomyopathy. Biochem Mol Biol Int 1996; 40: 487–495.
Dumoulin R, Sagnol I, Ferlin T, Bozon D, Stepien G, Mousson B : A novel Gly290Asp mitochondrial cytochrome b mutation linked to a complex III deficiency in progressive exercise intolerance. Mol Cell Probes 1996; 10: 389–391.
Andreu AL, Bruno C, Shanske S et al: Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology 1998; 51: 1444–1447.
Andreu AL, Bruno C, Dunne TC et al: A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol 1999; 45: 127–130.
De Lonlay P, Valnot I, Barrientos A et al: Mutations in BCS1 a mitochondrial respiratory chain assembly gene are responsive for the complex III deficiency of patients with tubulopathy, encephalopathy and liver failure. Nat Genet 2001; 29: 57–60.
Visapaa I, Fellman V, Vesa J et al: GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by point mutation in BCS1L. Am J Hum Genet 2002; 71: 863–876.
Haut S, Brivet M, Touati G et al: A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycemia and lactic acidosis. Human Genet 2003; 113: 118–122.
Rustin P, Chretien D, Bourgeron T et al: Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 1994; 228: 35–51.
Anderson S, Bankier AT, Barrel BH et al: Sequence and organisation of the human mitochondrial genome. Nature 1981; 290: 457–465.
MITOMAP Human mitochondrial genome database: Center for Molecular Medicine, Emory University, Atlanta, GA, USA (http://www.gen.emory.edu/mitmap.html).
Bouzidi MF, Carrier H, Godinot C : Antimycin resistance and ubiquinol cytochrome c reductase instability associated with a human cytochrome b mutation. BBA 1996; 1317: 199–209.
Keightley JA, Anitori R, Burton MD : Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am J Hum Genet 2000; 67: 1400–1410.
Degli Esposti M, De Vries S, Crimi M, Ghelli A, Patarnello T, Meyer A : Mitochondrial cytochrome b: evolution and structure of the protein. Biochim Biophys Acta 1993; 1143: 243–271.
Andreu AL, Hanna MG, Reichmann H et al: Exercice intolerance due to mutations in the cyt b gene of mitochondrial DNA. N Engl J Med 1999; 341: 1037–1044.
Papadimitriou A, Neustein HB, DiMauro S, Stanton R, Bresolin N : Histiocytoid cardiomyopathy of infancy: deficiency of reductible cytochrome b in heart mitochondria. Pediatr Res 1984; 18: 1023–1028.
Andreu AL, Checcarelli N, Iwata S, Shanske S, DiMauro S : A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr Res 2000; 48: 311–314.
Saint-Georges Y, Bonnefoy N, di Ragon JP, Chiron S, Dujardin G : A pathogenic cytochrome b mutation reveals new interactions between subunits of the mitochondrial bc1 complex. J Biol Chem 2002; 277: 49397–49402.
Johns DR, Neufeld MJ : Cytochrome b mutations in Leber hereditary optic neuropathy. Biochem Biophys Res Commun 1991; 181: 1358–1364.
Tawata M, Ohtaka M, Iwase E, Ikegishi Y, Aida K, Onaya T : New mitochondrial DNA homoplasmic mutations associated with Japanese patients with type 2 diabetes. Diabetes 1998; 47: 276–277.
Guan MX, Fischel-Ghodsian N, Attardi G : Nuclear background determines biochemical phenotype in deafness-associated mitochondrial 12SrRNA mutation. Hum Mol Genet 2001; 10: 573–580.
McFarland R, Clark KM, Taylor RW, Macphail S, Lightowlers RN, Turnbull DL : Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 2002; 30: 145–146.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haut, S., Villemeur, T., Brivet, M. et al. The deleterious G15498A mutation in mitochondrial DNA-encoded cytochrome b may remain clinically silent in homoplasmic carriers. Eur J Hum Genet 12, 220–224 (2004). https://doi.org/10.1038/sj.ejhg.5201132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201132
