
Abstract
We have identified a female patient with a complex phenotype that includes complete agenesis of the corpus callosum, bilateral periventricular nodular heterotopia, and bilateral chorioretinal and iris colobomas. Karyotype analysis revealed an apparently balanced, reciprocal, de novo chromosome translocation t(2;9)(p24;q32). Physical mapping of the translocation breakpoint by fluorescence in situ hybridization and PCR analysis led to the identification of two novel, ubiquitously expressed, Zn-finger-encoding transcripts that are disrupted in this patient. Unexpectedly, the rearrangement produced in-frame reciprocal fusion transcripts, making genotype–phenotype correlation difficult.
Similar content being viewed by others


Introduction
Agenesis of the corpus callosum (ACC) is one of the most common brain malformations found in humans, although the incidence is not known with certainty. Epidemiological data show a rate of 0.25 per 1000 live-births based on data collected primarily from newborns, which thus likely represents a significant underascertainment (Birth Defects and Genetic Diseases Branch, Metropolitan Atlanta Congenital Defects Program, Centers for Disease Control and Prevention, Atlanta, GA, 2001). ACC has been associated with many clinical and pathological findings including developmental delay, mental retardation, learning disabilities, infantile spasms and other seizure types, and many different congenital anomalies including other brain malformations.1 It is causally heterogeneous, having been associated with mutations in numerous identified genes, chromosome rearrangements, and metabolic disorders in both humans and mouse.2 Mutations in the FGFR2, CBP, XNP, L1CAM, MID1, and ARX genes result in syndromes that include ACC as part of the phenotype.3 Murine models of ACC include disruption of the MARCKS, bAPP, Vax1, Nfia, and Arx genes.3,4,5,6,7,8 These studies suggest that the ACC phenotype can result from several developmental disruptions including (1) primary malformations of the forebrain such as frontal encephaloceles, (2) absence of commisural neurons or axons due to abnormal cortical neuronal development, (3) disruption of the lamina terminalis (massa commissuralis) or related trophic molecules in the midline field, and (4) degeneration of the corpus callosum, usually triggered by the toxic effects of a metabolic disorder, although this is extremely rare.2 Over 150 human syndromes have been reported with ACC, implying that a very large number of genes contribute to callosal development. To date, however, very few human ACC-causing gene mutations have been identified.
We have identified a female patient with a balanced reciprocal, de novo chromosome translocation t(2;9) (p24;q32). The neurological phenotype of this patient, including ACC, suggests that the chromosomal rearrangement disrupts a gene(s) important for CNS development. Thus, experiments were initiated to define the molecular nature of the translocation. Two, novel ubiquitously expressed, Zn-finger genes were found to be disrupted by the chromosomal rearrangement, generating reciprocal in-frame fusion transcripts from each derivative chromosome. Our study suggests that both genes may be involved in CNS development. In addition, both genes now represent new candidate genes for ACC in other patients.
Materials and methods
BAC DNA and FISH analysis
STS content was confirmed for each BAC clone used for fluorescence in situ hybridization (FISH) analysis by PCR using a BioRAD icycler machine with the following PCR conditions: small numbers of bacteria containing each clone were used directly in a 25 μl PCR reaction with 5 μl 5 × cresyl red loading dye (0.05% cresyl red in 30% sucrose), 2.5 μl 10 × PCR buffer (0.67 mM Tris pH 8.8, 0.16 mM (NH4)2S04, 0.65 mM MgCl2, 0.5% Triton X-100 and 1% β-mercaptoethanol), 100 ng of each STS primer, 2 μl 2.5 mM dNTP, trace α-32P dATP, and 0.05 U Perkin-Elmer AmpliTaq. Cycling conditions in a BioRad iCycler were as follows: one cycle 94°C, 4 min, 35 cycles of 94°C 30 s, 55°C 30 s, 72°C 30 s, followed by one cycle of 72°C for 7 min. BAC DNA was then isolated for FISH analysis using an Autogen 740. Probes were directly labeled by nick translation with either Spectrum Orange-dUTP or Spectrum Green-dUTP (Vysis Inc., Downers Grove, IL, USA). Hybridizations were carried out according to previously published methods.9 Briefly, slides were washed for 2 min in 0.4 × SSC/0.3% NP-40 at 73°C, two minutes in 2 × SSC/0.1% NP-40 at room temperature, and a final wash of 4 min in 4X SSC/0.1% Tween-20 at room temperature. The slides were then mounted in anti-fade (Vector) with DAPI. The images were captured using a Zeiss Axiophot microscope with an #83000 filter (DAPI, FITC, and rhodamine) and a cooled CCD camera (Nu 200; Roper Scientific, Tucson, AZ, USA), and results were analyzed with SmartCapture 2 software (Digital Scientific).
Somatic cell hybrid analysis and patient lymphoblast RT-PCR
Somatic cell hybrids from patient lymphoblasts were generated by GMP Genetics and screened for chromosome 2 markers (D2S2952, D2S2944, and D2S434) and chromosome 9 markers (D9S169, D9S2169, and D9S1838) using similar PCR conditions to those described above. To generate cDNA, mRNA was extracted from lymphoblast cell lines of the patient and normal controls via Quiagen RNeasy midi column. Concentration of mRNA was then standardized using a TD-700 Fluorometer, and cDNA generated using Superscript II reverse transcriptase (Gibco). PCR conditions to amplify cDNAs were similar to those listed above. Primers used to confirm transcripts were as follows: For KIAA1803; 5F:GAGCATGCCTACAAGTGTTC, 5R:TGGAGTCTTCCACTTCTTCA, 8F:GGAGTAGTTTTCCGCTGTG, 8R:GAAGGTGGCTGTCCAGTT, 7F:CAGCATTTGGTATGTTAACG, 7R:AGAGACTCTTCCCTCCTTTTGAA. For ASXL2; 10F:AGCAGTGATGGTGGCAGCAAC, 10R:CTGGACCAACCTGTCGATC, 11R:GCTGTGCTTCTGTGATGG, 12R:CACTGGCTGCTTTGACAGTC. In some cases, PCR fragments were directly TOPO-cloned into the pCRII-TOPO vector (Invitrogen) and sequenced by the University of Chicago Sequencing Core Facility, using an ABI 3100 capillary sequencing machine.
Electronic-database information
Marshfield map: http://research.marshfieldclinic.org/genetics/
UCSC Human Genome Working Draft: http://genome.ucsc.edu/
Ensembl: http://www.ensembl.org/
Genbank, Link and AceView, UniSTS: http://www.ncbi.nlm.nih.gov/
GenBank accession numbers: D2S2952 [UniSTS:19840], D2S2944 [UniSTS:68648], D2S434 [UniSTS:60315], D9S169 [UniSTS:11939], D9S2169 [UniSTS:11939], and D9S1838 [UniSTS:9019], RPCI11- 404P12 [AC010150], RPCI11-63C2 [AC0648475], RPCI11-508N12 [AL512593], SHGC-86024 [G61621], SHGC-14156 [G16289] KIAA1803 gene [partial sequence AB058706, full-length mRNA XM_040560], hypothetical protein DKFZp762N2316 [CAB94868], Mus. Musculus gene-trap clone 4-2 [X66902], Homo sapiens IMAGE cDNA clone 2975237 [AW628946], clone 5297392 [BI561402], KIAA1685 [AB051472], dbest ID# 7481122 [BF908773], Mus Musculus IMAGE cDNA clone 2749639 [AW496276], clone 931044 [AI507091], Drosophila melanogaster Asx [AJ001164], Homo sapiens additional sex combs like 1, ASXL1 [AJ438952].
Results and discussion
Clinical phenotype
We studied a female with a complex phenotype that includes ACC together with bilateral chorioretinal and iris colobomas and bilateral periventricular nodular heterotopia (PNH), who was found to have a de novo, apparently balanced reciprocal translocation between chromosomes 2 and 9 with breakpoints at p24 and q32, respectively (Figure 1). She was born after a pregnancy complicated by gestational diabetes. Initial examination showed frontal bossing, left ptosis, dysplastic left ear, hemangiomas of the right forehead, left knee and shoulder and right vulva, bilateral leukocoria, hearing loss, ventricular septal defects, vesicoureteral reflux, and hyperreflexia of all four extremities. An eye examination detected chorioretinal and iris colobomas, and brain MRI demonstrated bilateral PNH and ACC (Figure 1). Mild concentric left ventricular hypertrophy was attributed to the maternal diabetes. The syndrome presented by this patient is novel and most likely represents primary ACC caused by abnormal CNS development, especially since it appears in the context of the additional developmental brain abnormalities including PNH and ocular coloboma. Analysis of the molecular nature of the translocation therefore is likely to lead to the identification of additional genes required for CNS development.

Clinical phenotype. (a) Sagittal MRI of an age-matched normal compared to (b) the patient's MRI at 13 months of age, demonstrating a complete absence of the corpus callosum. (c) Axial brain MRI demonstrating bilateral PNH. Arrows indicate foci of neuronal cell bodies along the lateral ventricles that did not migrate into the cerebral cortex during development. (d) Partial karyotype indicating the balanced translocation t(2;9)(p24;q32).
Mapping the breakpoints
To identify the genes potentially disrupted in this patient, we first mapped the translocation breakpoints using FISH. BAC clones from the RPCI-11 human library containing STS markers corresponding to relative locations on the Marshfield genetics map from chromosomes 2 and 9 were used as FISH probes on metaphase chromosome spreads from an Epstein–Barr virus-transformed lymphoblast cell line generated from the patient's peripheral blood. Using the relative STS positions on the Marshfield genetics map, BAC clones were chosen to cross the relevant regions on chromosomes 2 and 9. FISH analysis of each clone was then used to identify clones that mapped proximal or distal to each translocation breakpoint. In this manner, physical maps of chromosomes 2p24 and 9q31–q32 were generated and the breakpoint regions narrowed and defined. Figure 2 demonstrates that BAC 404P12 maps distal to the chromosome 2 breakpoint, while BAC 63C2 maps proximal to the breakpoint. Since the sequence of both BACs is complete, we determined that these two BACs overlap by approximately 2.4 kb, thus localizing the chromosome 2 breakpoint to very near or within this small region. Figure 3 summarizes the localization of the chromosome 9 breakpoint. FISH signals for BAC 508N12 were observed on the derivative 2 and 9 chromosomes as well as the wild-type chromosome 9 (Figure 3a, b), localizing the chromosome 9 breakpoint to a position within this BAC.

Chromosome 2 breakpoint analysis. (a) FISH analysis of patient metaphase chromosomes using BAC 63C2 (red) and chr. 9 alpha-satellite DNA (green). This BAC is proximal to the translocation breakpoint hybridizing to both the wild-type and derivative chromosome 2. In contrast (b), FISH analysis using BAC 404P12 (red), revealed only one signal on the wild-type chromosome 2 with the other located on derivative chromosome 9. Thus, 404P12 maps distal to the breakpoint, localizing the breakpoint to within or very near to this small region of overlap in these two BACs. (c) Physical map of an approximately 30 Mb region of chromosome 2p24. BAC names are listed above the lines and the approximate sizes in kilobase are below.

Chromosome 9 breakpoint analysis (a) Metaphase FISH analysis using BAC 508N12 (red) and chromosome 2 alpha-satellite DNA (green) probes. Three signals are observed with BAC 508N12 indicating that it spans the translocation breakpoint. (b) Interphase FISH analysis demonstrates three signals with probe 508N12 (red). Two chromosome 2 alpha-satellite DNA signals (green) are also present. (c) Physical map of an approximately 23 Mb region of chromosome 9q31–32.
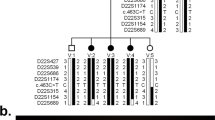
To further refine the location of the breakpoints within the identified BACs, somatic cell hybrids were constructed to isolate each derivative chromosome (GMP Genetics Inc.). The human chromosomal complement of each hybrid was determined by STS analysis using PCR. Three hybrids containing the derivative chromosome 2 were identified, as were two hybrids containing the derivative chromosome 9. STS markers contained within each putative breakpoint BAC were next systematically tested to locate each breakpoint. This analysis allowed us to narrow the chromosome 9 breakpoint to a 30 kb region within BAC 508N12, between primer pairs 5F/5R and 7F/7R (Figure 4a,c and data not shown). This analysis also confirmed that the chromosome 2 breakpoint was located between SHGC-86024 and SHGC-14156, both within the 2.4 kb region of overlap between BACs 404P12 and 63C2.

Gene structure of endogenous and fusion genes. (a) KIAA1803 contains 11 exons encompassing 84 kb of genomic DNA and encodes a protein with six C2H2-type Zn2+ finger domains marked by *. ASXL2 is defined by at least 13 exons covering 138 kb of genomic DNA and is represented by ESTs 2975237 [AW628946], 5297392 [BI561402], and KIAA1685 [AB051472]. The exon indicated by # has been reported only in clone 5297392 [BI561402], isolated from a testes library. EST sequences from other cDNA sources do not contain this exon, including clone dbest ID# 7481122 [BF908773]. Locations of PCR primers used to confirm the presence of transcripts in lymphoblast cDNA are indicated by small horizontal arrows in both the genes. (b) Fusion transcripts are illustrated with the breakpoints of each gene represented by a vertical arrow. The exact breakpoint of ASXL2 is within the genomic sequences 5′ to the # alternate exon, an exon not found in RNA from either normal or patient lymphoblast cells. (c) PCR analysis of somatic cell hybrids containing the derivative chromosome 2 and derivative 9 cell lines. Distal exon 7 of KIAA1803 are present in control genomic DNA, but not found in the derivative 9-containing hybrid. Instead, these exons are now associated with derivative 2-containing hybrids. In contrast, proximal exon 5 of KIAA1803 is present in both control genomic DNA and derivative 9 containing hybrids and is not found in the derivative 2 line, indicating that the KIAA1803 breakpoint is located between exons 5 and 7 of this gene. (d) PCR products from cDNA isolated from both normal (lanes 1,3,5,7,9,11) and patient (2,4,6,8,10,12) lymphoblasts for the indicated primer pairs. While the expected wild-type fragments were amplified for the endogenous genes in both normal and patient cDNA, the fusion transcripts are only detected in patient samples. The small bands in lane 5 are PCR artefacts associated with the use of primer 10F and are also present in lanes 1 and 2. The presence of these small bands in lane 5 is not indicative of a fusion transcript in the normal patient sample.
The translocation disrupts genes on both chromosomes 2 and 9
Once genomic breakpoint regions were identified, we constructed transcript maps of the breakpoint BACs from public human genome databases, compiling data from GenBank, Locus Link, UCSC Human Genome Working Draft, and Ensembl to identify disrupted genes that had potential to cause the complex phenotype of our patient.
The KIAA1803 gene on chromosome 9
The 30 kb region between 5F and 7R on chromosome 9 corresponds to a region inclusive of exons 5–7 of the KIAA1803 gene (Figure 4a). KIAA1803 was first identified as a large protein-encoding brain-expressed cDNA.10 The KIAA1803 gene is predicted to encode hypothetical protein DKFZp762N2316, represented by 11 exons across approximately 83 kb of genomic DNA encoding 6 C2H2-type Zn-fingers, two nuclear localization domains and a putative ER-membrane domain (AceView KIAA1803). KIAA1803 is the human homologue of mouse Gt4-2 (gene-trap clone 4-2). Gt4-2 was first identified in early gene trap experiments in mouse ES cells.11 Preliminary analysis indicated that homozygous mutant mice for the trapped allele of Gt4-2 either died at an undetermined time during gestation or survived with an associated growth retardation phenotype dependent upon the genetic background of the mutation. Heterozygous mice were also significantly smaller than wild-type littermates for undetermined reasons. The CNS phenotype of Gt4-2 mice was not examined and unfortunately the mice are now extinct, with ES cells no longer available. Additional mutant animals have not been generated. Both published and our own in situ data demonstrate that mouse Gt4-2 is ubiquitously expressed throughout the CNS from e9.5 to e15.5 encompassing the period when callosal development is initiated11.
The ASXL2 gene on chromosome 2
The small region of overlap between BACs 404P12 and 63C2 is located within an intron of a gene corresponding to dbEST id# clone 7481122 [BF908773]. BLAST analysis of this clone indicated considerable homology to the 5′ end of the Human Additional sex combs like (ASXL1) gene. Further, BLAST analysis revealed that this EST overlapped with another EST, dbest id# 2975237, again with considerable homology to both the 5′ region of the Drosophila Asx gene and the Human ASXL1 gene. Clones mapped on the Human Genome Working Draft (April 2001 freeze, May 2002 draft assembly) indicated that two other EST clones (clones 5297392 and KIAA1685) mapped to the region, both with homology to the 3′ end of the Drosophila Asx gene.12 Importantly, KIAA1685 appears to encode a conserved PHD Zn-finger domain at the 3' end, diagnostic for the Asx gene family (CL Fisher and HW Brock, in press). PCR analysis of human lymphoblast cDNA with primers crossing the ends of the ESTs demonstrated that all of these ESTs are indeed linked and represent one transcriptional unit encompassing approximately 138 kb of genomic DNA (Figure 4a and data not shown).
In Drosophila, the Asx gene encodes a chromatin protein that binds to Polycomb group sites on polytene chromosomes. Further, Drosophila Asx interacts genetically with the Polycomb and super sex combs genes and mediates repression or activation of homeotic genes.12,13 A human Asx-related gene (ASXL1) has recently been described, mapping to chromosome 20q11, with an exon structure similar to that which we have predicted for the combined ESTs at this locus. Based on transcript structure and homologies (NCBI, tBLASTx) to both Drosophila Asx and Human ASXL1 (CL Fisher and HW Brock, in press), we have determined that this locus on human chromosome 2 represents a new member of the Asx-like gene family. We have therefore named this gene ASXL2. To determine if the ASXL2 gene is expressed during CNS development and could contribute to the neurological phenotype of our patient, we identified two mouse Asxl2 encoding EST clones (2749639 and 931044) and performed whole-mount in situ hybridization during mouse embryogenesis.14 Similar to the Gt4-2 gene, mouse Asxl2 is also ubiquitously expressed throughout the CNS from e9.5 to e15.5 (data not shown).
Although nothing is known about the function of ASXL2, studies have begun to address the role of the related Asxl1 gene in the mouse.6 This analysis may serve to form a hypothesis regarding the role of ASXL2.
It is interesting to note that several homeobox genes are required for normal CNS development and may be targets of ASXL regulation. Specifically, Pax6, Lhx2, Crx, and Rex have all been implicated in various stages of eye development.15,16,17 ARX is another homeobox gene expressed during cortical development. Although missense mutations in the ARX gene have been demonstrated recently to cause X-linked infantile spasms, a severe form of epilepsy with onset in infancy,18,19 more severe mutations of ARX cause X-linked lissencephaly with absent corpus callosum and abnormal genitalia (XLAG) or isolated agenesis of the corpus callosum,3 which overlaps with the phenotype observed in our t(2;9) patient. Thus, it is tempting to speculate that at least ARX may be a candidate target gene for regulation by the ASXL2 protein.
Fusion transcript
Since database analysis of the two disrupted genes in this translocation patient indicated that both are transcribed in the same direction with respect to each other, there was a distinct possibility that fusion transcripts were generated in the translocation patient. To test this hypothesis, primers located near the breakpoint in each gene were chosen to amplify across the breakpoint region. As shown in Figure 4d, wild-type KIAA1803 and ASXL2 transcripts were detected in both normal and patient lymphoblast cDNA. In contrast, fusion transcripts were detected only in the patient samples. To confirm that these PCR products actually represent fusion transcripts, the amplified products were cloned and sequenced. This analysis confirmed that the products indeed represent chimeric transcripts derived from both a KIAA1803:ASXL2 fusion transcript and the reciprocal ASXL2:KIAA1803 fusion transcript. A summary of these data is illustrated in Figure 4. This sequence analysis also served to further localize the KIAA1803 breakpoint to the intron between the fifth and sixth exons. The ASXL2 gene breakpoint was localized to the intron after the fourth exon, next to an alternately spliced exon (# in Figure 4a and b), which was not found in either normal or patient lymphoblast RNA.
Sequence analysis of the fusion transcripts allowed us to make one additional and remarkable conclusion: the translocation event in our patient juxtaposed exons from each gene that are in frame with respect to each other in both chimeric transcripts. Thus, both fusion transcripts encode open-reading frames across the respective breakpoints which are contiguous with the open-reading frames of the primary genes (data not shown). Since antibodies are not available to detect either the KIAA1803 protein or the ASXL2 protein, we are unable to determine if reciprocal fusion proteins are also translated from the two fusion transcripts in this patient, however, this remains an intriguing possibility.
Summary
Our molecular analysis of the translocation t(2;9)(p24;q32) patient has demonstrated that although the translocation is itself a relatively simple, balanced translocation, the underlying molecular basis of the patient phenotype is actually very complex. Without further analysis of gene function in mouse models, we cannot differentiate if the patient phenotype results from loss of function of one allele of either KIAA1803 or ASXL2 or both in combination. Further, it is possible that the phenotype is caused by gain of function of either or both of the fusion gene products. It is also possible that the phenotype results from a combination of all or some of these molecular changes. Importantly however, our data have identified two novel candidate genes, KIAA1803 and ASXL2, likely to participate in corpus callosum and CNS development. It remains to be determined if either gene is a candidate gene for other sporadic cases of ACC. Clearly, these genes merit further functional analysis.
References
Lacey DJ : Agenesis of the corpus callosum. Clinical features in 40 children. Am J Dis Children 1985; 139: 953–955.
Dobyns WB : Absence makes the search grow longer. Am J Hum Genet 1996; 58: 7–16.
Kitamura K, Yanazawa M, Sugiyama N et al: Mutations of Arx/ARX cause abnormal development of forebrain and testes in mice, and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002; 32: 359–369.
Bertuzzi S, Hindges R, Mui SH, O'Leary DD, Lemke G : The homeodomain protein vax1 is required for axon guidance and major tract formation in the developing forebrain. Genes Dev 1999; 13: 3092–3105.
das Neves L, Duchala CS, Tolentino-Silva F et al: Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc Natl Acad Sci USA 1999; 96: 11946–11951.
Fisher C, Helgason, C, Bodner C et al: Targted disruption of a mouse homologue of the Drosphila Asx gene leads to bidirectional axial skeleton transformations and spermatocyte defects. Dev Biol 2002; 247: 458.
Muller U, Cristina N, Li ZW et al: Mice homozygous for a modified beta-amyloid precursor protein (beta APP) gene show impaired behavior and high incidence of agenesis of the corpus callosum. Ann NY Acad Sci 1996; 777: 65–73.
Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ : MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci USA 1995; 92: 944–948.
Chong SS, Pack SD, Roschke AV et al: A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 1997; 6: 147–155.
Nagase T, Nakayama M, Nakajima D, Kikuno R, Ohara O : Prediction of the coding sequences of unidentified human genes. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 2001; 8: 85–95.
Skarnes WC, Auerbach BA, Joyner AL : A gene trap approach in mouse embryonic stem cells: the lacZ reported is activated by splicing, reflects endogenous gene expression, and is mutagenic in mice. Genes Dev 1992; 6: 903–918.
Sinclair DA, Milne TA, Hodgson JW et al: The Additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development 1998; 125: 1207–1216.
Milne TA, Sinclair DA, Brock HW : The additional sex combs gene of Drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol Gen Genet 1999; 261: 753–761.
Fukuchi-Shimogori T, Grove EA : Neocortex patterning by the secreted signaling molecule FGF8. Science 2001; 294: 1071–1074.
Lupo G, Andreazzoli M, Gestri G, Liu Y, He RQ, Barsacchi G : Homeobox genes in the genetic control of eye development. Int J Dev Biol 2000; 44: 627–636.
Mathers PH, Jamrich M : Regulation of eye formation by the Rx and pax6 homeobox genes. Cell Mol Life Sci 2000; 57: 186–194.
Porter FD, Drago J, Xu Y et al: Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development 1997; 124: 2935–2944.
Bienvenu T, Poirier K, Friocourt G et al: ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002; 11: 981–991.
Stromme P, Mangelsdorf ME, Shaw MA et al: Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet 2002; 30: 441–445.
Acknowledgements
We are grateful to CL Fisher and HW Brock for sharing unpublished data with us and we thank the University of Chicago Clinical Cytogenetics laboratory for cell lines and G-banding. This study was supported by NIH Grant P01 NS39404 to WBD and DHL, Medical Scientist National Research Service Award Grant No. PHS 5 T 32 GM07281 to IG and a Brain Research Foundation grant to KJM.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first three authors contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ramocki, M., Dowling, J., Grinberg, I. et al. Reciprocal fusion transcripts of two novel Zn-finger genes in a female with absence of the corpus callosum, ocular colobomas and a balanced translocation between chromosomes 2p24 and 9q32. Eur J Hum Genet 11, 527–534 (2003). https://doi.org/10.1038/sj.ejhg.5200995
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200995
Keywords
This article is cited by
-
Periventricular heterotopia with complete agenesis of the corpus callosum
Journal of Neurology (2006)
