
Abstract
Neural tube defects (NTD) are among the most common congenital malformations in humans. The current view is that there are no major genes causing NTDs, but combinations of sequence variants in different genes have additive effects on determining the malformation. Therefore it is important to identify such sequence variants to get a better understanding of NTD pathogenesis. Studies on animal models have shown that BMP4 and NOG are involved in the patterning of the neural tube. We therefore performed a single-strand conformation analysis (SSCA) mutation screen for both genes in 179 spina bifida aperta (SBA) patients. Our SSCA screen revealed four missense mutations in BMP4 and one in NOG. It is likely that these mutations have acted together with other gene variants in independently segregating loci as susceptibility factors in these SBA cases. In addition, a case-control association study provides evidence for a genotype disequilibrium of BMP4 polymorphism 455T→C (V152A) in exon 5. The frequency of the heterozygous 455TC genotype is lower in cases than in controls (nominal P=0.017), although allele frequencies are similar in both groups. A possible explanation for this finding might be that BMP4 455TC heterozygosity at this site is a protective factor in the normal population, although this hypothesis cannot be proven to date.
Similar content being viewed by others

Introduction
Neural tube defects (NTD) are among the most frequent congenital malformations in humans. The current hypothesis is that they are of multifactorial origin due to the combined effects of sequence variants in several genes and environmental factors.1 Identifying such sequence variants should provide a better understanding of NTD pathogenesis.
The various phenotypes of NTD are thought to arise from an impaired primary neurulation process at different levels of the body axis. Since studies on neural tube formation in human embryos are rare, the current knowledge of this developmental process is mainly based on data from model organisms.2 Expression studies on chicken or mouse embryos at the time of neural tube formation provide information on genes relevant in neurulation (neurulation genes). Since many of these developmental genes are highly conserved during evolution, they may also contribute to neural tube formation in man.
Numerous mouse strains (spontaneous and targeted mutant mice) with congenital malformations including NTDs have been described and the responsible genes have been listed as potential candidate genes for human NTD.3,4 Not all of these mutated genes have an impact on the neurulation process, but they show adverse effects on embryonic morphogenesis either during the time of neurulation or post-neurulation thus leading to a neural tube defect (NTD genes).5 Therefore, looking for genes causing NTDs in humans it might be best to choose those genes that are expressed during neural tube formation (neurulation genes) and are causing NTDs in animal models.
The secreted polypeptides Bmp4 and Noggin were chosen from the candidate lists as both are neurulation genes: Bmp4 is expressed in the dorsal epidermal ectoderm, posterior mesoderm and the presumptive neural crest cells.6 Noggin transcripts are found in notochord and dorsal neural tube.7 Both are NTD genes since targeted inactivation in mice leads to abnormal phenotypes: Bmp4 knockout mice exhibit little mesoderm differentiation causing disorganised structures (including a small neural plate and cranially open folds); mice lacking Noggin show skeletal abnormalities and defects in growth and patterning of the neural tube.7,8 Haploinsufficiency for NOG has been shown to cause proximal symphalangism (MIM 185800), whereas up to now no human disease has been associated with BMP4 mutations. Our study with spina bifida aperta (SBA) patients revealed single point mutations in both genes that may have acted as susceptibility factors in the individual cases. Interestingly, an association study provides evidence for a genotype disequilibrium between SBA patients and controls for the BMP4 polymorphism 455T→C (V152A). The frequency of the heterozygous 455T→C genotype is lower in cases than in controls. A possible explanation is that BMP4 heterozygosity at this site is a protective factor in the normal population, although this hypothesis cannot be proven to date.
Probands and controls
A total of 179 unrelated German children and young adults (83 females, 96 males) with non-syndromic spina bifida aperta (meningocele and meningomyelocele) ascertained through Spina Bifida Clinics were included in the study. Eleven spina bifida aperta (SBA) cases were familial with at least one affected first-degree family member. All other patients were the only affected in their families. The control population consisted of 161 unrelated healthy German students and young adults (51 males, 110 females) without a family history of NTD. The study design was approved by the local ethics committee of the University of Marburg. Informed consent was obtained from patients, their parents and controls.
BMP4 and NOG mutation analysis
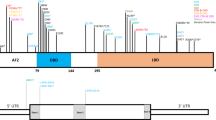
To screen BMP4 for mutations (GenBank accession no. NM_001202, AL138479) the two coding exons (exon 4 and 5) including the exon-intron boundaries were amplified by PCR with optimised product sizes for single strand conformation analysis (SSCA). The genomic sequence of NOG was retrieved from GenBank accession no. U31202 and overlapping primer pairs were designed to cover the coding sequence in the single exon (Table 1). SSCA and allele-specific sequencing of aberrant and normal SSCA conformers were performed as described earlier.9
BMP4 polymorphism
Patients and controls were genotyped by SSCA for the previously reported 455T→C (V152A) polymorphism in exon 5 using primer pair 3f/3r (Table 1, Figure 1).10 The resulting SSCA genotypes were confirmed in five patients and five controls by two independent methods: restriction digestion of the PCR products with HphI and by direct sequencing of the PCR products.

SSCA pattern (10°C) of BMP4 455T→C polymorphism in exon 5 (A); family pedigree of SBA patient 80168 (B) displaying an aberrant SSCA pattern due to the mutation 272C→G in exon 4. Genotypes: −/− homozygous wildtype, +/− heterozygous; aberrant bands are indicated by arrowheads.
Statistics
To investigate differences in allele and genotype distributions between patients and controls the two-sided Pearson's χ2 test for 2×2 tables was performed at a 5% significance level. For deviation from Hardy-Weinberg equilibrium in the two groups we used the one-sample χ2 test for goodness-of-fit with one degree of freedom (software package StatXact-3, Version 3.1).
Results
BMP4
A case-control study for 179 patients and 161 controls revealed no differences in BMP4 455T→C allele frequencies (Table 2). Further exploratory analysis showed a difference in 455T→C genotype frequencies. 455TC heterozygotes were significantly more frequent in controls (59.0%) than in cases (45.8%; nominal P=0.017). 455TT and 455CC homozygosity did not differ between cases and controls (P=0.20 and P=0.11 respectively). When testing genotype frequencies of both groups for Hardy-Weinberg equilibrium a significant deviation with an excess of 455TC heterozygotes was found in controls (P=0.01), but not in cases (P=0.40).
In female patient 80168 a heterozygous missense mutation was found in exon 4 (Table 3, Figure 1). A cytosine is replaced by a guanine at position 272 (272C→G), changing the predicted serine residue at position 91 to cysteine (S91C). The same mutation was found in the patient's unaffected mother, brother and in one healthy control individual. A missense mutation was detected in female patient 80246. The nucleotide transversion from adenosine to guanine at position 673 (673A→G) in exon 5 leads to an amino acid change from threonine to alanine at position 225 (T225A). The patient's mother and brother were heterozygous carriers of the mutation, but none of the tested 161 control individuals. Male patient 80201 showed a close-by heterozygous missense mutation at nt 676 (676C→T) which produces an arginine to tryptophan amino acid substitution (R226W). The mutation was also found in the unaffected father and in one control individual. In male patient 80495 the substitution of guanine with cytosine at position 1100 (1100C→G) introduced a serine to threonine exchange S367T, which was inherited from the mother. None of the 161 controls showed this alteration.
NOG
In female patient 80332 a missense mutation was detected (275G→A) leading to a glycine to glutamic acid exchange (G92E). The mutation was also present in her unaffected father and in one control individual. Neither in patients nor controls were any polymorphisms found in the coding region (Table 3).
Discussion
Findings in NTD knock-out models led us to the conclusion that Bmp4 and Noggin play an important role in neurulation. Therefore mutations in BMP4 and NOG, in combination with other genetic or environmental factors, could result in human neural tube malformations.
SSCA screening of BMP4 coding sequences revealed four heterozygous missense mutations in four different SBA patients. All sequence variants were inherited from an unaffected heterozygous parent. Three missense mutations (S91C, T225A, R226W) are in the BMP4 pro-peptide region responsible for dimerization of the protein. Although the pro-peptide is cleaved from the rest of the protein before secretion, experiments have shown that sequence alterations in this region influence the stability of the mature protein.11 One missense mutation (S367T) was found within the C-terminal physiologically active part of the protein and therefore might have had a direct impact on the function of the mature protein. In summary, the sequence variants in our SBA cases could have caused subtle changes in protein stability/activity and therefore contributed to the development of the malformation.
The NOG SSCA screen revealed no polymorphisms and one missense mutation (G92E) in a SBA patient. The replaced glycine is within a stretch of five glycines and is located in the variable region 1 of the protein.12 This region contains seven amino acids that are highly conserved in mammals and are essential for proper structure and function of the protein.
Since the BMP4 and NOG mutations were also found in unaffected individuals the sequence alterations alone are not sufficient to induce the patients' SBA. Taking into account the hypothesis of a polygenic nature of NTD, the SBA phenotype has to be explained by concurrence of the described mutations with additional sequence variants in independently segregating loci.
The case-control association study of the BMP4 polymorphism revealed that genotype distributions differ between SBA patients and controls, although allele frequencies are similar in both groups. However, only the difference between the 455TC genotype frequencies reaches nominal significance. The 455TC frequency is lower in cases than in controls (nominal P=0.017). To evaluate whether cases or controls are in Hardy–Weinberg disequilibrium we compared the observed and expected genotype frequencies within each group. The controls deviate from Hardy–Weinberg equilibrium. (P=0.01), due to an increased heterozygous 455TC genotype frequency in controls. In the original polymorphism report of Mangino et al, 1999 no disequilibrium was observed, but the number of investigated Caucasian individuals was low (n=30).
To date possible effects of this polymorphism on structure and processing of the BMP4 pro-peptide are unknown. But since an obvious shift towards heterozygosity is observed in the control population, the 455TC genotype might be an advantage for these individuals by preventing the formation of NTD during embryogenesis. From our data we cannot decide whether the 455TT or 455CC genotype might have acted as susceptibility factors for SBA in the tested population. We cannot finally exclude that the Hardy-Weinberg disequilibrium is due to selection bias. Therefore, further association studies are necessary to confirm the deviation in genotype distribution on a wider scale in other populations. Potential effects of the observed mutations on the development of SBA have to be elucidated in functional studies.
References
Elwood JM, Little J, Elwood JH . Epidemiology and Control of Neural Tube Defects Oxford: Oxford University Press 1992
Copp A, Cogram P, Fleming A et al. Neurulation and neural tube closure defects Methods Mol Biol 2000 136: 135–160
Juriloff DM, Harris MJ . Mouse models for neural tube closure defects Hum Mol Genet 2000 9: 993–1000
Shimeld SM . The evolution of dorsoventral pattern formation in the chordate neural tube Am Zool 1999 39: 641–649
Colas JF, Schoenwolf GC . Towards a cellular and molecular understanding of neurulation Dev Dyn 2001 221: 117–145
Dudley AT, Robertson EJ . Overlapping expression domains of bone morphogenetic protein family members potentially account for limited tissue defects in BMP7 deficient embryos Dev Dyn 1997 208: 349–362
McMahon JA, Takada S, Zimmerman LB, Fan C-M, Harland RM, McMahon AP . Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite Genes Dev 1998 12: 1438–1452
Winnier G, Blessing M, Labosky PA, Hogan BLM . Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse Genes Dev 1995 9: 2105–2116
Stegmann K, Boecker J, Richter B et al. A screen for mutations in human homologues of mice exencephaly genes Tfap2α and Msx2 in patients with neural tube defects Teratology 2001 63: 167–175
Mangino M, Torrente I, De Luca A, Sanchez O, Dallapiccola B, Novelli G . A single-nucleotide polymorphism in the human bone morphogenetic protein-4 (BMP4) gene J Hum Gen 1999 44: 76–77
Constam DB, Robertson EJ . Regulation of bone morphogenetic protein activity by pro domains and proprotein convertases J Cell Biol 1999 144: 139–149
Valenzuela DM, Economides AN, Rojas E et al. Identification of mammalian Noggin and its expression in the adult nervous system J Neurosci 1995 15: 6077–6084
Acknowledgements
We would like to thank the patients and their families for co-operating in this study. We thank Christopher Schwarz for technical assistance. The work was supported by the Deutsche Forschungsgemeinschaft, Bonn, the organization ARQUE, Mainz and Stiftung PE Kempkes, Marburg.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Felder, B., Stegmann, K., Schultealbert, A. et al. Evaluation of BMP4 and its specific inhibitor NOG as candidates in human neural tube defects (NTDs). Eur J Hum Genet 10, 753–756 (2002). https://doi.org/10.1038/sj.ejhg.5200875
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200875
Keywords
This article is cited by
-
Current perspectives on the genetic causes of neural tube defects
Neurogenetics (2006)
