
Abstract
Mutations in the Cx26 (GJB2) gene have been shown to be responsible for a major part of autosomal recessive non-syndromic inherited prelingual deafness. We have sequenced the coding region of GJB2 gene from 169 Taiwanese patients with prelingual deafness and 100 unrelated normal individuals. In the deaf patients, three mutations were found: two novel mutations, 551G→A, and 299-300delAT, and one previously described mutation, 235delC. Four previously reported polymorphisms, 79G→A, 109G→A, 341A→G, and 608T→C, were also found in both deaf patients and normal individuals and one new possible polymorphism, 558G→A, which was only found in a patient. Interestingly, we did not find the 35delG allele, which is commonly found in the Caucasian population, either in the patients or in normal individuals we examined. Our data also showed 235delC to be the most common type of mutation found in Cx26 mutants (approximately 57%). Therefore, based on our findings, we have developed a simple molecular test for the 235delC mutation and it should be of considerable help to those families to understand the cause of their children having the prelingual deafness.
Similar content being viewed by others


Introduction
The most common inherited sensory disorder is severe hearing impairment, which affects about 1 in 1000 children.1 Currently, over 40 chromosomal loci have been identified for non-syndromic hearing loss through linkage studies (http://www.uia.ac.be/dnalab/hhh/). However, only 22 cloned nuclear genes have been shown so far to cause non-syndromic deafness (http://www.uia.ac.be/dnalab/hhh/hhhgenes.html), and nearly one half of the cases of non-syndromic deafness among Caucasian and Japanese are reported to be caused by mutations of Cx26 gene.2,3,4 Cx26 (GJB2) encodes connexin 26 protein which is a member of the connexin protein family and can interact with connexin 32, connexin 46, and connexin 50 to form a hexameric of homotypic (composed of six identical connexin subunits) or heterotypic (composed of more than one species of connexins) half channel (connexon) of gap junctions. Such gap junctions play an important role in the local circulation of potassium ions in the inner ear.5, 6 Connexin 26 has been reported to be expressed in the stria vascularis, basement membrane, limbus and spiral prominence of cochlea.7 Mutations in Cx26 have also been found to be involved in various mechanisms that lead to the loss of hearing, such as interference with the proper oligomerisation or intracellular transport of Cx26, impairment of interaction between connexons in opposing cells, or block the recycling of potassium ions back to the endolymph of the cochlear duct after stimulation of the sensory hair cells.8
There is a lot of evidence indicating that mutations of Cx26 might be a major contributor to prelingual deafness among Caucasians9,10,11,12 and Japanese.4 However, there have been significant differences in the types and frequencies of mutation of Cx26 found between the two populations. 35delG has been reported to be the most common form of Cx26 mutation among Caucasians, but 235delC is reported to be the most frequently found form of Cx26 found among Japanese. We analysed the coding region of Cx26 of 169 Taiwanese schoolchildren with prelingual hearing loss. Of the 169 children, 19 were having mutations in this region. Of these 19, eight were found to be homozygous for 235delC, two heterozygous for 235delC, four compound heterozygous for 235delC/299-300delAT, two heterozygous for 299-300delAT, one heterozygous 551G→A, and two heterozygous for 558G→A. Our data identified 235delC to be the most common type of mutation found in Cx26 mutants (∼57%).
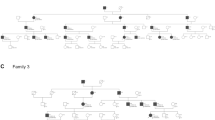
Family studies were also performed on nine probands with Cx26 mutations, eight of whom were shown to have inherited mutant allele(s) from the parentsrelatives. The results of this study further illustrate the contribution of Cx26 mutants to the pathogenesis of prelingual hearing loss in children.
Materials and methods
The present investigation, a connexin 26 analysis, was performed on children from the National Taichung School for the Deaf. A complete medical history was obtained from each child to record the age of onset of deafness and to exclude the possibility of such environmental causes as maternofoetal infection, perinatal complications, meningitis, mumps, prenatal or postnatal drug ototoxicity, and acoustic trauma. One hundred and sixty-nine children (103 males, 66 females), between 4 and 18 years of age had mild to profound prelingually sensorineural deafness. Family studies were performed on nine probands with Cx26 mutation. One hundred randomly selected normal individuals were screened to be used as control subjects.
Using a QIAamp DNA Blood Kit (Qiagen), we obtained DNA samples from 5 ml of peripheral blood from each individual. The quality and quantity of purified genomic DNA were determined by running a 0.8% agarose gel and spectrophotometry.
The primer pair used in polymerase chain reaction and two additional internal primers used in DNA sequencing were: Cx26-AU (5′-TCTTTTCCAGAGCAAACCGC-3′), forward primer for PCR and sequencing; Cx873-L (5′-CTGGGCAATGCGTTAAACTGG-3′), reverse primer for PCR and sequencing; Cx501F (5′-GTGGCCTACCGGAGACATGAG-3′), primer for internal forward sequencing; AL (5′- GGACACGAAGATCGCTGCAG-3′), primer for internal reverse sequencing. The coding region of Cx26 was PCR amplified by denaturation of template DNA at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 60°C for 1 min, extension at 72°C for 1 min, and finally at 72°C for 5 min. The PCR products were purified using a PCR Purification Kit (Qiagen). The purified DNA were then subjected to PCR-directed DNA sequencing using a DNA Sequencing Kit (Perkin-Elmer) and ABI Prism 310 Genetic Analyzer (Perkin-Elmer).
To verify the 235delC mutation, we used ApaI to digest the PCR product in a final volume of 20 μl: 5 μl of PCR product, 5 units of ApaI, 2 μl of 10X buffer, and 0.2 μl of 100X BSA. The reaction were carried out at 25°C for 2 h, and then run on a 2% agarose gel.
Results
The Cx26 mutations observed in Taiwanese patients with prelingual non-syndromic hearing loss were two deletions, 235delC and 299-300delAT, and one substitution, 551G→A. In addition to the 235delC mutation, two other mutations were found to be novel. The prevalence of Cx26 mutations are summarised in Table 1, and the effects of each mutation as well as the mutant allele frequency are summarised in Table 2. Our data also identified that 235delC is the most common mutant allele found in Cx26. However, we did not find any heterozygotes for mutant alleles among the 100 controls. Of the four 235delC/299-300delAT compound heterozygote deaf schoolchildren, two were sisters (assigned 043 and 053). Further sequence analysis of this F043/053 family, revealed the father to be heterozygous for 299-300delAT/wt and the mother heterozygous for 235delC/wt. However, both parents had normal hearing levels, indicating that the novel 299-300delAT mutation acts in a recessive manner. It is worth noting that the 35delG allele, which is common in Caucasian population, was not found in either the deaf patients or normal controls. We also studied eight families (14 family members in total) to learn more about the inheritance of individual mutations. It was found that all of the deaf children in these family families inherited the recessive mutant alleles from both parents except for patient 129, who had only one Cx26 mutant allele and whose alleged father had a wild-type homozygote for Cx26.
Four polymorphic sites of Cx26 were determined in the 100 unrelated normal controls as well as in our patient population (data not shown). The 341A→G and 608T→GC polymorphisms have been previously described by Kudo et al.4 and the other two polymorphisms, 79G→A and 109G→A, have been previously described by Kelley et al.13 In addition to these known polymorphisms, one new possible polymorphism, 558G→A, was detected in one patient (see discussion below).
Discussion
We have found three mutations, 551G→A, 299-300delAT and 235delC, in the Cx26 gene among Taiwanese patients with prelingual deafness. Among those the 235delC mutation is the allele with the highest frequency in the Taiwanese deaf population. Similarly, the 235delC mutation has been described previously by Kudo et al. as being the most frequent allele found in Japanese deaf people,4 though it has not been reported in the Caucasian population. Of the 169 deaf schoolchildren we studied, two had 235delC/wt heterozygotes. Since this mutation has been characterised as recessive4 (and our data), it is very likely that these patients may have had other mutation(s) in other gene(s) which are responsible for the clinical manifestation of deafness. The 299-300delAT causes a frameshift and produces a premature Cx26 that stops at position 113. Patients 043 and 053 are siblings with the same genotype of 299-300delAT/235delC. Their father is 299-300delAT/wt heterozygote and their mother is 235delC/wt heterozygote. Both parents showed no hearing impairment. According to the results we obtained by studying the F043/053 family, this 299-300delAT mutation exhibited the characteristics of autosomal recessive heredity. Thus, the two 299-300delAT/wt heterozygote deafness school children, like 235delC/wt carrier, may also have had other mutation(s) in other genes responsible for deafness. Patient 129 was heterozygous for 551G→A/wt. Although his father was also deaf, the father did not show any mutation in Cx26. The mother of patient 129 was a normal individual but we could not obtain her DNA for sequence analysis. In patient 129's family (F129), the hearing loss of the father was clearly not caused by the mutation in Cx26 and the child may have either inherited the other hearing loss factor(s) from the father and demonstrated the phenotype or perhaps due to the de novo 551G→A allele which caused the disease. Because the 551G→A is a missense mutation that causes R184Q substitution in Cx26 and the amino acid is located at the second extracellular loop, this mutation might affect the selective compatibility between different species of connexin proteins to form heterotypic functional channels.14 However, whether the 551G→A is sufficient cause for the development of deafness or not needs further study. Another substitution found in this study was 558G→A. This substitution does not change the amino acid composition of Cx26 (silent mutation). However, this substitution was found in patient 117 only, whose parents showed no hearing impairment. Unfortunately, we could not obtain his parents' DNA to confirm whether he was a case of de novo mutation or whether there was mutation in another gene responsible for this phenotype. From the data we obtained, this silent mutation may represent a polymorphism rather than a pathogenic mutation. However, there is increasing evidence that the silent mutation might cause phenotypic variability by influencing splicing accuracy or efficiency.15 Thus, we can not rule out the possibility that 558G→A may has the pathogenic effect. Further study is currently underway to verify this view point.
It is very interesting that we did not find any 35delG mutation in either our patients or in our controls. The 35delG allele has been demonstrated to be the most frequently found mutation to cause non-syndromic recessive prelingual deafness in Caucasians.9,16 Tekin et al16 suggested that the high frequency of 35delG allele may possibly arise from a founder effect and further enhanced by assortative mating in the population. This postulation could be true, since the oriental populations, Japanese4 and Taiwanese (this study), do not exhibit this allele and the most common mutation in both oriental populations is 235delC, indicating that different causes between the Cx26 mutations of Caucasian and those of oriental populations.
One specific mutation, 235delC, accounted for the majority (approximately 57%) of Cx26 mutant alleles in our study. Adding our findings to those of the Japanese study, we can say that 235delC is one of the most frequent disease mutations identified among oriental populations to date. Genetic counselling for prelingual deafness has been so far considerably hampered by the difficulty in distinguishing genetic and non-genetic deafness in families presenting with a single child. Based on the results presented in this study, we used restriction enzyme ApaI to verify the 235delC mutation and for the quick screening for such mutation. Since 235delC eliminates an ApaI site, therefore, we can identify an individual who is a normal, heterozygote, or homozygote for this mutation by ApaI digestion of the PCR product.
References
Morton NE . Genetic epidemiology of hearing impairment Ann NY Acad Sci 1991 630: 16–31
Zelante L, Gasparini P, Estivill X et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans Hum Mol Genet 1997 6: 1605–1609
Cohn ES, Kelley PM . Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss Am J Med Genet 1999 89: 130–136
Kudo T, Ikeda K, Kure S et al. Novel Mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population Am J Med Genet 2000 90: 141–145
Salt AN, Melichar I, Thalmann R . Mechanisms of endocochlear potential generation by stria vascularis Laryngoscope 1987 97: 984–991
Salt AN, DeMott JE . Ionic and potential changes of the endolymphatic sac induced by endolymph volume changes Hear Res 2000 149: 46–54
Kelsell DP, Dunlop J, Stevens HP et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness Nature 1997 387: 80–83
White TW . Functional analysis of human Cx26 mutations associated with deafness Brain Res Rev 2000 32: 181–183
Denoyelle F, Weil D, Maw MA et al. Prelingual deafness: high prevalence of a 30delG 30delG mutation in the connexin 26 gene Hum Mol Genet 1997 6: 2173–2177
Gasparini P, Estivill X, Volpini V et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families Eur J Hum Genet 1997 5: 83–88
Guilford P, Ben Arab S, Blanchard S et al. A non-syndromic form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q Nature Genet 1994 6: 24–28
Maw MA, Allen-Powell DR, Goodey RJ et al. The contribution of the DFNB1 locus to neurosensory deafness in a Caucasian population Am J Hum Genet 1995 57: 629–635
Kelley PM, Harris DJ, Comer BC et al. Novel mutations in the connexin 26 gene (IGJB2) that cause autosomal recessive (DFNB1) hearing loss Am J Hum Genet 1998 62: 792–799
Krutovskikh V, Yamasaki H . Connexin gene mutations in human genetic diseases Mut Res 2000 462: 197–207
Cartegni L, Chew SL, Krainer AR . Listening to silence and understanding nonsense: exonic mutations that affect splicing Nat Rev Genet 2002 3: 285–298
Tekin M, Akar N, Cin S et al. Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35delG mutation in Caucasians Hum Genet 2001 108: 385–389
Acknowledgements
We thank Kuei-Hsiang Hung, Chung-Hsiang Yang, Yu-Che Cheng, Shi-Hsin Huang, and Chien-Hsun Wu for their technical assistance and to National Health Research Institute for support to CCL (NHRI-Ex90-8933SL). This work is supported by National Science Council, Republic of China (NSC 89-2745-P-040-002, NSC 89-2320-B-040-061, and NSC 90-2320-B-040-032).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, YC., Kung, CY., Su, MC. et al. Mutations of Cx26 gene (GJB2) for prelingual deafness in Taiwan. Eur J Hum Genet 10, 495–498 (2002). https://doi.org/10.1038/sj.ejhg.5200838
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200838
Keywords
This article is cited by
-
A novel method for detecting nine hotspot mutations of deafness genes in one tube
Scientific Reports (2024)
-
Rare compound heterozygosity involving dominant and recessive mutations of GJB2 gene in an assortative mating hearing impaired Indian family
European Archives of Oto-Rhino-Laryngology (2017)
-
Dew inspired breathing-based detection of genetic point mutation visualized by naked eye
Scientific Reports (2014)
-
Lower carrier rate of GJB2 W24X ancestral Indian mutation in Roma samples from Hungary: implication for public health intervention
Molecular Biology Reports (2014)
-
Genetic mutations in nonsyndromic deafness patients of Chinese minority and han ethnicities in Yunnan, China
Journal of Translational Medicine (2013)
