
Abstract
Adult stem cells hold great promise for future therapeutic applications. Hematopoietic stem cells (HSCs) are among the best-characterized adult stem cells. As such, these cells provide a conceptual framework for the study of adult stem cells from other organs. Here, we review the current knowledge of HSC generation during embryonic development and HSC maintenance in the bone marrow (BM) during adult life. Recent scientific progress has demonstrated that the development of HSCs involves many anatomical sites in the embryo, but the relative contribution of each of these sites to the adult HSC pool remains controversial. Specialized anatomical sites in the BM have been identified as stem cell niches, and these play essential roles in regulating the self-renewal and differentiation of HSCs through recently identified signaling pathways. Extracellular signaling from stem cell niches must integrate with the intracellular molecular machinery and/or genetic programs to regulate HSC fate choice. The exact cellular and/or molecular mechanisms defining stem cell niche and ‘stemness’ of HSC is largely unknown although substantial progress has been made recently. Hence, many questions remain to be answered even in this relatively well-defined model of stem cell biology.
Similar content being viewed by others

Main
Hematopoiesis is a term used to describe the process of blood cell formation during both the embryonic and adult stages of an organism. Hematopoiesis is also viewed as the process of development, self-renewal and differentiation of hematopoietic stem cells (HSCs), a type of adult stem cell that is the source of all blood cell lineages. In fetal and adult mammals, HSCs predominantly reside in fetal liver (FL) and bone marrow (BM), respectively. However, HSCs do not originate in FL or BM, but rather migrate from other tissues to these sites during embryonic development. In mammals, most blood cells have relatively short lifespans. For this reason, HSCs continuously differentiate into multiple lineages of different blood cell types, simultaneously replicating themselves through self-renewal to prevent depletion of the stem cell pool in the BM. The marrow contains specialized environments that regulate the balance of HSC self-renewal and differentiation and comprise what has been termed the stem cell niche. With recent developments in the fields of multicolor flow cytometry and high-speed cell sorting, inducible gene targeting and advanced imaging techniques, we have gained a wealth of knowledge about the cellular and molecular mechanisms underlying the development, self-renewal and differentiation of HSCs. One of the ultimate goals of current research is to recapitulate this complex process using in vitro cell-culture systems, which will facilitate the translation of new discoveries into therapeutic benefits.
Development of HSCs: Lessons Learned from Developmental Embryology
In mice, hematopoietic cells are among the first cell types to emerge during embryogenesis.1 Originating from mesodermal precursors, embryonic hematopoiesis generates differentiated blood cells to meet the immediate demands of embryonic growth as well as the HSCs that form the foundation of adult hematopoiesis. Interestingly, embryonic hematopoiesis involves multiple anatomical sites (the extraembryonic yolk sac (YS), the intraembryonic aorta–gonad–mesonephros (AGM) region, the placenta, the thymus and the FL), most of which are not involved in adult hematopoiesis. HSCs arise and migrate between these multiple sites until the BM develops sufficiently to provide the environmental niches necessary for HSC function. The FL is the predominant site of hematopoiesis from embryonic day 12 (E12) through birth, after being seeded by HSCs that migrate from other sites through the circulation.2 HSCs undergo dramatic expansion and differentiation in the FL before migrating into the BM (Figure 1).

The different anatomical sites involved in embryonic hematopoiesis and their proposed relationships. A subset of mesodermal cells commits to the hematopoietic and endothelial lineages through a bipotent progenitor termed the hemangioblast. These hemangioblasts probably migrate into the yolk sac (YS), aorta–gonad–mesonephros (AGM) and placental regions to give rise to primitive and definitive hematopoiesis. Hematopoietic stem cells (HSCs) may be generated in some or all of the three sites before they migrate to fetal liver (FL) for expansion. FL HSCs migrate to bome marrow (BM) at around the time of birth
Gastrulation in the mouse embryo leads to the formation of ectoderm, mesoderm and endoderm from the epiblast. As gastrulation progresses, extraembryonic and intraembryonic regions are separated as YS and embryo proper. Morphologic evidence of embryonic hematopoiesis is first observed in the YS. The precursors that give rise to YS hematopoiesis migrate from mesoderm of the posterior regions of the primitive streak.1 These mesodermal precursors give rise to both hematopoietic and endothelial cells in the YS. Primitive (visceral) endoderm cells that contact with mesodermal precursors in the YS play important instructive roles in specifying hematopoietic and endothelial lineages by secreting soluble factors.3 Shortly after gastrulation at E7.0–7.5, multiple foci of red spots called the ‘blood islands,’ which consist of clusters of hematopoietic cells surrounded by a flattened endothelium, appear on the surface of the YS.4 Primitive erythroid cells, the first blood cells formed within the blood islands, are large, nucleated red blood cells resembling the erythrocytes of lower vertebrates. These cells, which eventually enucleate after entering the circulation,5 contain specialized isoforms of hemoglobin that function optimally in the embryonic environment. The erythroid progenitors that give rise to primitive red blood cells can be cultured and will form colonies of erythroid cells in vitro; these primitive erythroid colony-forming cells (CFC) are termed EryP-CFC. EryP-CFC activity is detected in the YS as early as E7.25 and is extinguished by E9.0.1 Primitive erythrocytes enter the blood vessels from the YS at the onset of circulation (∼8.5 d.p.c.). The early YS also contains primitive macrophages and megakaryocytes that are slightly different from their adult counterparts.6, 7 Interestingly, these cells are derived from monopotent progenitors rather than multipotent progenitors. At later stages of development, the YS contains mast-cell progenitors, granulocyte–macrophage progenitors and definitive erythroid progenitors.1 Whether the myeloid progenitors are derived from the same precursors as the primitive erythroid cells is currently unknown, but a bipotent erythroid–megakaryocyte progenitor has recently been characterized in the YS.8 The later stage myeloid and erythroid progenitors do not differentiate into mature cells within the YS, but rather seed the FL once the circulation system is established at around E8.5.1 After the establishment of circulation, cells with both myeloid and lymphoid potential as well as cells with long-term repopulating potential can be detected in the YS, but whether these cells are generated in situ or migrate to the YS from other sites is unknown.
Previous studies have suggested that the YS was the sole site of de novo generation of hematopoietic cells from mesodermal precursors, including the HSCs that seed the FL.9, 10, 11 This view was challenged through a series of elegant chicken–quail chimeric experiments, in which quail intraembryonic compartments were grafted onto chicken YS prior to initiation of circulation between the two sites (hence ruling out a possible cross-contamination by circulating hematopoietic cells). These experiments demonstrated that only quail cells contributed to long-term multilineage hematopoiesis.12 Similar results were also reported in Xenopus developmental studies.13 These experiments established that in avian and amphibian species, the YS provides only transient hematopoiesis, mainly consisting of primitive erythroid and myeloid cells, while the embryo proper is the major site for the generation of definitive HSCs for adult hematopoiesis.
Hematopoietic progenitor generation has also been demonstrated in murine intraembryonic regions, raising the possibility that, as in birds and frogs, mammalian hematopoiesis may arise independently in both intraembryonic and extraembryonic sites.14, 15, 16 The para-aortic splanchnopleura (pSP) region in mouse develops from the intraembryonic lateral plate mesoderm contacting the endoderm. After E9.5, the pSP gives rise to AGM region. While intraembryonic hematopoiesis has been identified in the pSP/AGM region,17 difficulties inherent in tracking development during mammalian embryogenesis, including the developmental site (intrauterine rather than in an easily accessible egg), the small size of the tissues of interest, the rapid rate of developmental events, the onset of circulation at about the same time as hematopoiesis during mouse development (E8–9), and the need to stage accurately separate embryos by somite counts have resulted in controversy over whether hematopoiesis originates simultaneously in both intraembryonic and extraembryonic sites, or whether mammalian intraembryonic hematopoiesis is seeded by circulating cells that originate in the YS. The origin of definitive HSCs for adult hematopoiesis is also highly disputed.
Early experiments to address these questions relied on taking the embryonic tissues out of their original environment and putting them into artificial in vitro differentiating assays and/or in vivo repopulation assays using radiation- or drug-conditioned fetal or adult animals. From these experiments, three distinct views of early mouse hematopoiesis have surfaced. In one set of studies, the pSP and YS tissues were isolated before the onset of circulation and the explants cultured for 2 days before transplanting the cells into irradiated adult recipient mice. The pSP progenitors could generate long-term multilineage engraftment, while the YS progenitors generated only short-term erythro-myeloid reconstitution.17 Thus, explant cultures using tissues obtained prior to the onset of circulation suggest that mammalian hematopoiesis mirrors what is seen in avian and amphibian models, in that the sole source of definitive HSC is intraembryonic and that the YS functions only in a transient manner to provide a limited degree of blood development. However, the isolation of cells from their natural environment and culturing them in a non-physiological assay system may have altered their developmental pathways. Hence, these experiments do not conclusively rule out the possibility of a YS contribution to adult hematopoiesis in vivo.
In contrast to this work, recent studies have shown that the interruption of either the onset of circulation by targeted disruption of Ncx1, or of engraftment of hematopoiesis by targeted disruption of Rac1, resulted in normal YS hematopoiesis, but a failure in the development of intraembryonic hematopoiesis.18, 19 These targeted deletion models suggest that the YS is the sole source of hematopoietic function, and that all other sites are seeded from the YS. This view is also supported by older data from a number of investigators.9, 10 However, again, the use of artificial in vitro assays in these studies may not reveal the in vivo potential of these tissues completely. In addition, the effects of disruption of these genes on hematopoiesis, aside from the disruption of blood circulation and cell migration, are unclear. A third view holds that both the YS and the AGM region independently generate the definitive HSCs that ultimately seed the FL. Matsuoka et al.20 showed that co-culture of cells from either YS or pSP with an AGM-derived stromal cell line generated definitive HSCs, suggesting that both the YS and AGM cells contribute to definitive hematopoiesis. Furthermore, a quantitative assessment of HSC production at the beginning of FL hematopoiesis, and correlation with tissue distribution of progenitors, led a separate group of investigators to suggest that both the YS and AGM contribute to the burst of definitive HSCs found in the FL at E12.21
A more physiologically relevant way to address whether or not the YS contributes to adult hematopoiesis is by genetically labeling YS cells at their early stages before the establishment of circulation, and measuring their contribution to later stages of hematopoiesis in vivo. A recent study utilized the Cre/loxp recombination system to ask this question.22 A gene encoding a fusion protein containing the Cre recombinase gene flanked by two copies of a mutated estrogen receptor was placed under the transcriptional control of the Runx1 locus, which was demonstrated to be selectively expressed in the YS of the precirculation embryo. Nuclear localization of Cre was transiently induced by injection of tamoxifen, resulting in the activation of reporter gene expression in the Runx1-expressing YS cells and their progenies. By following the labeled cells during embryonic development and adult stages, the authors concluded that some YS-derived cells migrate through AGM, umbilical cord and FL, seed the BM, and contribute to adult hematopoiesis. This study supports the idea that both YS and AGM hematopoiesis contribute to the adult HSC pool in mice. However, several caveats raised in the experimental approach of this elegant study must be acknowledged. The generation of the reporter mouse strain involved the inactivation of one allele of the Runx1 gene, resulting in a change in gene dosage, which may have altered the developmental pathways of extraembryonic and/or intraembryonic hematopoiesis. Furthermore, although the authors demonstrated that expression of Runx1 is largely restricted to the YS in the precirculation embryo, it is possible that a few intraembryonic hematopoietic precursors also express Runx1 at E7.5. These few intraembryonic-labeled cells may have expanded and contributed to the later stages of hematopoiesis. In spite of these caveats, this study provides strong evidence in support of a YS origin of at least a subset of adult HSC.
Murine placentas were recently shown to contain HSC activity as early as E9.0.23, 24, 25 HSCs in placenta expanded dramatically between E11.5 and E12.5. As a result, the placenta has over 15-fold more HSCs than the AGM region and the YS. Currently, the origin of placental HSCs is unknown. They may be generated de novo within the placenta, or they may migrate from other anatomical sites such as the YS or AGM.
From these studies, a relatively clear picture of embryonic hematopoiesis emerges: the YS is the initial site of embryonic hematopoiesis, and mesodermal precursors give rise to both endothelial cells and hematopoietic cells in the YS. Hematopoiesis in the YS mainly produces primitive erythrocytes, primitive myeloid cells and some definitive myeloid progenitors, but also generates cells destined to persist as adult HSC. The intraembryonic AGM region is a major site of the in situ generation of definitive HSCs. The placenta may provide a suitable environment for HSC maturation and/or expansion, similar to the role played by the FL, before colonization of the marrow at around the time of birth. The relative contributions of YS, AGM and placenta to the FL and adult HSC pool is still unclear and awaits additional investigations. The inducible labeling model utilized by Samokhvalov et al.22 could be adapted to label hematopoietic precursors within specific embryonic sites to measure the contribution of these labeled cells to adult hematopoiesis.
Self-Renewal of HSCs
During embryonic development, HSCs generated in the AGM region, YS or placenta must colonize and expand in the FL to produce enough HSCs to ultimately seed the BM, where life-long hematopoiesis occurs. Throughout the adult life of a mammalian organism, HSCs must replicate themselves to maintain the constant HSC pool in the marrow. The process of HSC replication through mitosis is called self-renewal. Since FL HSCs are more actively engaged in cell division than are BM HSCs, the mechanisms that regulate HSC self-renewal are probably different between FL and BM. The fate choice of HSCs to either self-renew or differentiate is controlled by a complex interplay between intrinsic mechanisms and extrinsic signals from the surrounding environment or stem cell niches.26 Understanding the mechanisms of self-renewal of HSCs will facilitate the expansion of HSCs in culture while maintaining stem cell identity for cell and gene therapies.
Hematopoietic stem cell niches
FL and BM are the major organs for HSC expansion, maintenance and differentiation. The FL environment supports the rapid expansion of HSCs.27 HSCs must undergo symmetric cell divisions in FL, in which one HSC gives rise to two identical daughter HSCs. Efforts have been made to identify the key cellular and molecular elements of the FL microenvironment that support the rapid self-renewal of HSCs. One group of researchers has established numerous stromal cell lines from FL and compared their abilities to support self-renewal of HSCs in vitro, resulting in at least one cell line that can actually support self-renewal.28 The researchers also compared the gene expression profiles of HSC-supportive stromal cells with that of HSC-nonsupportive stromal cells, and have identified multiple differentially expressed genes.29 The functions of many of these genes in the regulation of HSC self-renewal have yet to be defined. Another group has identified a population of FL cells characterized as being positive for CD3 and negative for Ter119 that can support HSC self-renewal in culture.30 Although this population is quite heterogeneous, gene expression profile and functional studies have identified a number of secreted proteins, including insulin-like growth factor 2 and angiopoietin-like proteins, as the key HSC-supportive factors.30, 31
In adult BM, the HSC number stays relatively constant in the absence of overt injury or blood loss. Under normal conditions, most BM HSCs are quiescent (Figure 2). However, the majority of these cells divide regularly, as demonstrated by their slow, constant incorporation of nucleotide analogues.32, 33 The cell-cycle state of HSCs correlates to their multipotency, since most HSCs that are capable of long-term engraftment of irradiated recipients are in the G0 phase of the cell cycle.34 It is widely assumed that the dividing HSCs in BM undergo asymmetric cell division, in which an individual HSC gives rise to non-identical daughter cells, one keeping the HSC identity and the other becoming a differentiated progenitor cell. There are two hypothetical mechanisms by which asymmetric cell division might be achieved: divisional asymmetry and environmental asymmetry. In divisional asymmetry, specific cell-fate determinants in the genome or cytoplasm (RNA or proteins) are distributed unequally during cell division. After cell division, only one daughter cell receives the determinants, thus retaining the HSC fate while the other daughter differentiates. In environmental asymmetry, one HSC produces two identical daughter cells initially; however, only one remains in the HSC niche and retains the stem cell identity, while the other enters a different environment favoring its differentiation. A recent study provided evidence for divisional asymmetry in muscle stem cells by showing that the template strand of DNA was more often retained by cells fated to remain undifferentiated, while the newly synthesized DNA strand was more often found in cells that had acquired a more differentiated phenotype.35

Mouse hematopoietic stem cells (HSCs) and progenitor cells were isolated from adult bone marrow (BM) and characterized by differential interference contrast (DIC) microscopy, mitochondrial staining using a fluorescent probe (Rh123), and by transmission electron microscopy (TEM). HSCs are characterized by a relatively quiescent metabolic state as indicated by mitochondrial staining, while multipotent progenitor (MPP) cells are metabolically active
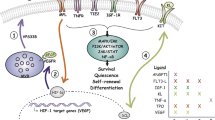
The concept of HSC niches was first proposed by Schofield.36 However, it has been only recently that the HSC niches in adult BM were directly observed and their components partially revealed.37 Currently, two types of HSC niches have been identified in BM; the endosteal niche located on the surface of trabecular bone, and the vascular niche at the BM sinusoids, which are low-pressure blood vessels with a fenestrated endothelium located in the center of BM. Osteoblasts are mesenchymal cells that produce the bone matrix to form the bone after mineralization. Osteoblasts are found on the endosteal surface lining between the bone and the marrow. Earlier studies suggested osteoblasts as candidates for a component of the HSC niche, to which transplanted HSCs home and reside.38 Osteoblast cell lines secrete many cytokines that promote the proliferation of hematopoietic cells in culture, and support the in vitro maintenance of HSCs.39 Direct evidence supporting osteoblasts as an essential component of the HSC niche comes from recent studies in which the osteoblast numbers were experimentally increased or decreased, which resulted in the HSC number changing in a parallel manner. In one of the studies, conditional knockout mice lacking the bone morphogenic protein receptor IA (BMPR1A) in the BM stroma (including osteoblasts) showed an increase in the number of both osteoblasts and long-term repopulating HSCs (LTR HSCs). In addition, the osteoblasts and LTR HSCs were shown to interact directly with each other through homotypic N-cadherin interactions.40 In another study, a transgenic mouse model was made to express receptors for parathyroid hormone and parathyroid hormone-related proteins under the control of the osteoblast specific, collagen-α1 promoter.41 This resulted in an increase in the number of both osteoblasts and HSCs in the marrow. The BM stromal cells derived from this transgenic mouse strain contained more osteoblasts and were more efficient in supporting in vitro maintenance of isolated BM HSCs than the stromal cells derived from normal mice. A third study showed that the conditional ablation of osteoblasts by the tissue-specific expression of thymidine kinase, which induces apoptosis in response to ganciclovir, caused a decrease in the number of HSCs in BM.42 These studies strongly suggest that osteoblasts residing on the bone surface are key components of the endosteal HSC niche (Figure 3).

Model showing two different hematopoietic stem cell (HSC) niches identified in the adult mouse bone marrow (BM). The endosteal niche is located at the surface of bone where quiescent HSCs contact with osteoblasts and CXCL12-abundant reticular (CAR) cells. Several of the molecules thought to be involved in endosteal niche function are shown. The vascular niche is located adjacent to the BM sinusoids. The HSCs in the vascular niche interact with CAR cells and possibly endothelial cells. Active self-renewal and differentiation of HSCs is probably more likely to happen in the vascular niche. HSCs in the vascular niche may also be mobilized into circulation. Under certain conditions, HSCs in the endosteal niche and the vascular niche may be exchangeable
Osteoblasts probably regulate HSCs through cell-surface adhesion molecules and/or secreted signaling molecules. It is essential to identify these molecules to understand fully the function of the HSC niche. Cell-adhesion molecules expressed by osteoblasts, such as N-cadherin and β1-integrin, are likely to be important in anchoring HSCs to the endosteal HSC niche.43 Osteoblasts also produce secreted and cell-surface-bound signaling ligands that bind to receptors on HSCs. Angiopoietin-1 (Ang-1) is one such ligand. In adult BM, Ang-1 is expressed primarily by osteoblasts while Tie2, the receptor for Ang-1, is expressed by HSCs. In vivo immunofluorescent imaging showed that these Tie2+ HSCs are localized to the bone surface and in contact with Ang-1-expressing osteoblasts.44 Moreover, Ang-1/Tie2 signaling is important in sustaining the quiescence and long-term repopulating activity of HSCs in adult BM.44, 45 Other signaling ligands produced by osteoblasts include the Notch ligand Jagged1, Wnt proteins and others.41, 46 One of the key characteristics of the endosteal stem cell niche is the high extracellular calcium-ion concentration ([Ca2+]).47 The HSCs express CaR, a G-protein-coupled receptor responding to the extracellular [Ca2+] level. Interestingly, a recent study showed that CaR-deficient mice had fewer HSCs in BM, and relatively more HSCs were found in the circulation and spleen. CaR−/− FL HSCs are less effective at repopulating irradiated transplant recipients, and this deficit could be attributed to a defect in localization to the endosteal stem cell niche after transplantation.47
It has been shown that a combination of signaling lymphocyte activation molecule (SLAM) expression defines long-term HSCs (CD150+CD48−CD244−) from adult BM using FACS.48 Because of its limited expression pattern, this combination of SLAMs can also be used in immunofluorescent imaging of LT-HSCs in tissue sections. By using this technique, another HSC niche has been identified in the sinusoidal endothelium. It has been shown that more cells with the LT-HSC phenotype are found attached to the sinusoidal endothelium within the center of BM than to the bone surface.48 The endothelial cells were initially proposed to act as the niche cells that interact with HSCs in this vascular niche.43 A subsequent study identified a type of reticular cell that expresses high levels of the chemokine CXCL12 (also known as stromal-derived factor 1, SDF-1) interacting with HSC interacting cells within the sinusoidal vascular niche. These reticular cells were named CXCL12-abundant reticular (CAR) cells. CXCL12–CXCR4 interactions are essential for the maintenance of the HSC pool in adult BM.49 CAR cells are present and interact with HSC in both the vascular and endosteal niches, which suggests a function linkage between these two niches. This finding suggests that there are probably several different types of cells functioning as HSC-supporting cells in both niches.
These observations also beg the question of why several different niches for HSCs exist, and what their specific roles in HSC self-renewal and differentiation might be. By using BrdU labeling, one study showed that BrdU+ long-term retaining HSCs are highly enriched at the bone surface compared to the center of marrow.40 This result suggests that HSCs residing in the endosteal niche are probably more quiescent than the HSCs residing in the vascular niche. This idea is reinforced by another study in which Ang-1 was shown to be expressed mainly by osteoblasts, and that Ang-1/Tie2 signaling slowed down the cell cycling of HSCs.44 To definitively clarify this issue, further studies comparing the repopulation activity, cell-cycle status and the gene expression profile of HSCs isolated from each of the stem cell niches are required.
Molecular mechanisms regulating HSC self-renewal.
External environmental signals must integrate with intrinsic molecular machinery to control the fate choices of individual HSC. Recently, a number of advances have been achieved in the search for a molecular signature to define the ‘stemness’ of stem cells. While very little is known about the actual mechanisms leading to the regulation of stemness, as more and more genetic pathways are uncovered, more candidates intimately involved in the regulation of HSC self-renewal will be identified (Table 1).
Currently, several transcription factors are implicated in the regulation of HSC self-renewal through various gain- and loss-of-function studies. One of them is the transcription factor translocation Ets leukemia (Tel), which has been demonstrated to be required for HSC self-renewal.55 Inactivation of Tel leads to the depletion of HSCs in BM without influencing their committed progenitors. Another interesting factor is HoxB4, a member of the homeobox gene family that encodes a set of transcription factors regulating embryonic body patterning and organogenesis. Overexpression of HoxB4 in HSCs has been shown to enhance the self-renewal of HSCs in both in vitro and in vivo models.56 Interestingly, HoxB4 knockout mice exhibit normal hematopoiesis, with only a mild defect in the proliferative potential of HSC.57 This discrepancy is likely due to the redundant function of other Hox family members. Growth factor independence 1 (Gfi1) is a zinc-finger-containing transcriptional repressor. Two studies independently identified Gfi1 as a positive regulator of HSC self-renewal that functions by restraining HSC proliferation.58, 59 Gfi1−/− HSCs exhibit increased proliferation and decreased capability for repopulation of irradiated recipients. It has been suggested that Gfi1 might function through upregulating the cell-cycle inhibitor p21, since p21 expression is greatly decreased in Gfi1−/− HSCs. Consistent with this hypothesis is the observation that p21-deficient HSCs also show impaired repopulation capability compared to wild-type HSCs.60 Another cell-cycle inhibitor, p18, has an opposite effect on HSC self-renewal compared to p21 such that p18-deficient HSCs have improved repopulation capability compared to wild-type HSCs.61, 62 These results suggest that a delicate balance of cell proliferation and cell-cycle repression is essential for HSC self-renewal. Stat3 and Stat5 are transcription factors involved in the JAK–STAT pathway that are implicated in the induction of leukemia, lymphoma and some solid tumors. Gain and loss of function studies have shown that both Stat5 and Stat3 are positive regulators of HSC self-renewal.63, 64
In addition to transcription factors, other proteins involved in the modulation of gene expression have been found to regulate self-renewal of HSCs. Bmi-1 is a polycomb group (PcG) protein that forms the Polycomb repression complex 1 (PRC1)with other members of the PcG family, such as Mel-18, Mph1/Rae28, M33, Scmh1 and Ring1A/B. This group of proteins represses the activity of the SWI-SNF chromatin-remodeling complex, leading to the repression of transcriptional activity. The Bmi-1 protein complex also binds to methylated histone H3 and contributes to the maintenance of epigenetic memory.65 Mph1/Rae28-deficient FL cells include fewer HSCs compared to wild-type mice.66 Bmi-1-deficient BM HSCs have profound defects in long-term repopulating activity,67 while forced expression of Bmi-1 in HSCs enhances the ex vivo expansion of HSCs and their in vivo repopulating activity.65 The mechanisms whereby Bmi-1 and its protein complex maintains HSC self-renewal are unclear, but it may be attributed to the repression of p16 and p19, and/or repression of differentiation-related gene expression programs.
Extrinsic environmental signals are connected to HSC self-renewal-specific gene expression programs by numerous signal transduction pathways. Notch, Wnt, BMP and Sonic hedgehog (Shh) signaling pathways have all been implicated in the regulation of HSC self-renewal. Notch proteins are highly conserved cell-surface receptors that include four members in mammals. The ligation of Notch ligands (five members in mammals) with Notch receptors results in cleavage and release of the intracellular domain of Notch receptors, which enters the nucleus and binds with the transcriptional repressor CSL, converting it into a transcriptional activator and inducing target gene expression. Notch signaling has important functions in cell-fate determination in many contexts of embryogenesis as well as in T-cell versus B-cell lymphoid lineage commitment. Forced activation of Notch1 in hematopoietic cells increases the self-renewal capability of HSCs and favors lymphoid lineage commitment.51 Notch signaling is active in HSCs and is downregulated as HSCs differentiate.52 One of the Notch ligands, Jagged1, is expressed at high level in osteoblasts, an essential component of HSC niches, suggesting that Notch signaling is important in controlling HSC self-renewal.41 Using a ‘pan’ Notch signaling inhibitor to inhibit Notch signaling in HSCs results in the accelerated differentiation and depletion of HSCs.52 In contrast to these findings, a recent study showed that the inactivation of Notch1 and/or its ligand Jagged1 in BM-hematopoietic cells and -stromal cells has no effect on HSC self-renewal. Notch1-deficient HSCs repopulated irradiated mice with Jagged1-deficient BM stroma normally, even under competition with wild-type HSCs.53 A possible explanation for this discrepancy is the functional redundancy of other members of the Notch family of receptors and/or ligands.
Wnt signaling has also been implicated in the regulation of HSC self-renewal. Like Notch signaling, Wnt signaling is highly conserved and acts during embryogenesis as well as in adult tissue homeostasis. The Wnt family comprises secreted proteins and includes many members. The receptors for Wnt proteins are the Frizzled family of seven-pass transmembrane proteins and LDL-receptor-related proteins.52 Wnt proteins can trigger at least three intracellular signaling pathways: the canonical β-catenin pathway, the non-canonical calcium pathway and the c-Jun N-terminal kinase pathway.68 β-Catenin is the central player of canonical Wnt pathway. In the absence of Wnt signaling, β-catenin is associated with a large protein complex called the destruction complex, which is targeted for degradation by ubiquitilation. Wnt protein binding to its receptor inhibits the degradation of β-catenin, resulting in the accumulation of β-catenin in the cytosol. β-Catenin then translocates into nucleus and binds with members of the LEF-TCF family of transcription factors, resulting in the expression of their target genes. Purified Wnt3A protein has been shown to promote self-renewal of HSCs.46 It has also been demonstrated that Wnt signaling is active in HSCs, and that the overexpression of an active form of β-catenin in HSCs leads to the expansion of immature cells that can reconstitute irradiated recipients.54 In contrast, HSCs transduced with inhibitors of Wnt signaling lost their in vivo repopulation capability. However, these experiments were performed using HSCs that expressed the antiapoptotic gene bcl-2, making the interpretation of the sole role of Wnt signaling in HSC self-renewal unclear. In contrast, another group showed that mice with conditionally deleted β-catenin had no hematopoietic defects, and that β-catenin-deficient HSCs had normal self-renewal capability.69 More recently, two independent studies addressed the role of canonical Wnt signaling in hematopoiesis by engineering mice that conditionally express dominantly active forms of β-catenin in hematopoietic cells. These mice showed widespread hematopoietic abnormalities and died within 2–3 weeks. The HSCs from these mice had lost the capacity to repopulate irradiated recipients, even though the number of cells expressing the HSC surface antigens increased.68, 70 Taken together, the existing studies suggest that canonical Wnt signaling may not be strictly required for HSC function, but that canonical Wnt signaling may affect self-renewal and differentiation of HSCs depending on the extent of canonical Wnt signaling and on the context of expression of additional genes. Non-canonical Wnt signaling and/or other signaling pathways may also compensate for the absence of canonical Wnt signaling in maintaining the self-renewal of HSCs.
Conclusion
After more than a half-century of scientific investigation and clinical practice of HSC biology, we are now entering a new era of science and medicine with respect to HSCs. Functional genomics and other newly developing technologies will continue to extend our understanding of the development, self-renewal and differentiation of HSCs. Deeper insight into the molecular and cellular mechanisms of HSC biology will ultimately lead to protocols for generation, expansion and control of differentiation of HSCs in vitro for clinical use in cell and gene therapies.
Abbreviations
- HSC:
-
hematopoietic stem cell
- YS:
-
yolk sac
- AGM:
-
aorta–gonad–mesonephros
- pSP:
-
para-aortic splanchnopleura
- FL:
-
fetal liver
- BM:
-
bone marrow
- CFC:
-
colony-forming cells
- CAR:
-
CXCL12-abundant reticular
- Tel:
-
translocation Ets leukemia
- Gfi1:
-
growth factor independence 1
- PcG:
-
polycomb group
- PRC1:
-
Polycomb repression complex 1
- BMP:
-
bone morphogenic protein
- Shh:
-
Sonic hedgehog
References
McGrath KE, Palis J . Hematopoiesis in the yolk sac: more than meets the eye. Exp Hematol 2005; 33: 1021–1028.
Johnson GR, Moore MAS . Role of stem cell migration in initiation of mouse fetal liver haematopoiesis. Nature 1975; 258: 726–728.
Belaoussoff M, Farrington SM, Baron MH . Hematopoietic induction and respecification of A-P identity by visceral endoderm signaling in the mouse embryo. Development 1998; 125: 5009–5018.
Lensch MW, Daley GQ . Origins of mammalian hematopoiesis: in vivo paradigms and in vitro models. Curr Topics Dev Biol 2004; 60: 127–196.
Kingsley PD, Malik J, Fantauzzo KA, Palis J . Yolk sac-derived primitive erythroblasts enucleate during mammalian embryogenesis. Blood 2004; 104: 19–25.
Palis J, Robertson S, Kennedy M, Wall C, Keller G . Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999; 126: 5073–5084.
Xu MJ, Matsuoka S, Yang FC, Ebihara Y, Manabe A, Tanaka R et al. Evidence for the presence of murine primitive megakaryocytopoiesis in the early yolk sac. Blood 2001; 97: 2016–2022.
Tober J, Koniski A, McGrath KE, Vemishetti R, Emerson R, de Mesy-Bentley KK et al. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007; 109: 1433–1441.
Moore MAS, Metcalf D . Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol 1970; 18: 279–296.
Weissman I, Papaioannou V, Gardner R . Fetal hematopoietic origins of the adult hematolymphoid system. In: Clarkson B (ed). Differentiation of Normal and Neoplastic Hematopoietic Cells. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 1978 pp 33–47.
Liu CP, Auerbach R . In vitro development of murine T cells from prethymic and preliver embryonic yolk sac hematopoietic stem cells. Development 1991; 113: 1315–1323.
Dieterlen-Lièvre F . On the origin of haemopoietic stem cells in the avian embryo: an experimental approach. J Embryol Exp Morphol 1975; 33: 607–619.
Chen XD, Turpen JB . Intraembryonic origin of hepatic hematopoiesis in Xenopus laevis. J Immunol 1995; 154: 2557–2567.
Medvinsky AL, Samoylina NL, Muller AM, Dzierzak EA . An early pre-liver intraembryonic source of CFU-S in the developing mouse. Nature 1993; 364: 64–67.
Cumano A, Furlonger C, Paige CJ . Differentiation and characterization of B-cell precursors detected in the yolk sac and embryo body of embryos beginning at the 10- to 12-somite stage. Proc Natl Acad Sci (USA) 1993; 90: 6429–6433.
Godin I, Dieterlen-Lievre F, Cumano A . Emergence of multipotent hemopoietic cells in the yolk sac and paraaortic splanchnopleura in mouse embryos, beginning at 8.5 days postcoitus. Proc Natl Acad Sci (USA) 1995; 92: 773–777.
Cumano A, Ferraz JC, Klaine M, Di Santo JP, Godin I . Intraembryonic, but not yolk sac hematopoietic precursors, isolated before circulation, provide long-term multilineage reconstitution. Immunity 2001; 15: 477–485.
Lux C, McGrath K, Conway S, Palis J, Yoder MC . Circulation plays an essential role in distributing mammalian yolk sac definitive hematopoietic progenitor cells to the embryo proper; Using the ncx1 knockout mouse model to prevent circulation. Blood 2005; 106: 154a.
Ghiaur G, Ferkowicz MJ, Bailey J, Yoder MC, Williams DA . Rac1 regulates migration of yolk sac hematopoietic progenitors into the blood, a process essential for subsequent seeding of fetal liver hematopoiesis. Blood 2005; 106: 154a.
Matsuoka S, Tsuji K, Hisakawa H, Xu M, Ebihara Y, Ishii T et al. Generation of definitive hematopoietic stem cells from murine early yolk sac and paraaortic splanchnopleures by aorta–gonad–mesonephros region-derived stromal cells. Blood 2001; 98: 6–12.
Kumaravelu P, Hook L, Morrison AM, Ure J, Zhao S, Zuyev S et al. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): role of the aorta–gonad–mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development 2002; 129: 4891–4899.
Samokhvalov IM, Samokhvalova NI, Nishikawa S . Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 2007; 446: 1056–1061.
Alvarez-Silva M, Belo-Diabangouaya P, Salaun J, Dieterlen-Lievre F . Mouse placenta is a major hematopoietic organ. Development 2003; 130: 5437–5444.
Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HK . The placenta is a niche for hematopoietic stem cells. Dev Cell 2005; 8: 365–375.
Ottersbach K, Dzierzak E . The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev Cell 2005; 8: 377–387.
Moore KA, Lemischka IR . Stem cells and their niches. Science 2006; 311: 1880–1885.
Ema H, Nakauchi H . Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood 2000; 95: 2284–2288.
Moore KA, Ema H, Lemischka IR . In vitro maintenance of highly purified, transplantable hematopoietic stem cells. Blood 1997; 89: 4337–4347.
Hackney JA, Charbord P, Brunk BP, Stoeckert CJ, Lemischka IR, Moore KA . A molecular profile of a hematopoietic stem cell niche. Proc Natl Acad Sci (USA) 2002; 99: 13061–13066.
Zhang CC, Lodish HF . Insulin-like growth factor 2 expressed in a novel fetal liver cell population is a growth factor for hematopoietic stem cells. Blood 2004; 103: 2513–2521.
Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med 2006; 12: 240–245.
Bradford GB, Williams B, Rossi R, Bertoncello I . Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol 1997; 25: 445–453.
Cheshier SH, Morrison SJ, Liao X, Weissman IL . In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci (USA) 1999; 96: 3120–3125.
Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL . Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med 2005; 202: 1599–1611.
Conboy MJ, Karasov AO, Rando TA . High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol 2007; 5: e102.
Schofield R . The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978; 4: 7–25.
Wilson A, Trumpp A . Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol 2006; 6: 93–106.
Askenasy N, Zorina T, Farkas DL, Shalit I . Transplanted hematopoietic cells seed in clusters in recipient bone marrow in vivo. Stem Cells 2002; 20: 301–310.
Taichman RS, Reilly MJ, Emerson SG . Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 1996; 87: 518–524.
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003; 425: 836–841.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003; 425: 841–846.
Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL . Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004; 103: 3258–3264.
Potocnik AJ, Brakebusch C, Fassler R . Fetal and adult hematopoietic stem cells require beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 2000; 12: 653–663.
Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004; 118: 149–161.
Puri MC, Bernstein A . Requirement for the TIE family of receptor tyrosine kinases in adult but not fetal hematopoiesis. Proc Natl Acad Sci USA 2003; 100: 12753–12758.
Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003; 423: 448–452.
Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006; 439: 599–603.
Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ . SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005; 121: 1109–1121.
Sugiyama T, Kohara H, Noda M, Nagasawa T . Maintenance of the hematopoietic stem cell pool by CXCL12–CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006; 25: 977–988.
Priestley GV, Scott LM, Ulyanova T, Papayannopoulou T . Lack of alpha4 integrin expression in stem cells restricts competitive function and self-renewal activity. Blood 2006; 107: 2959–2967.
Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT . Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 2002; 99: 2369–2378.
Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol 2005; 6: 314–322.
Mancini SJ, Mantei N, Dumortier A, Suter U, MacDonald HR, Radtke F . Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood 2005; 105: 2340–2342.
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003; 423: 409–414.
Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev 2004; 18: 2336–2341.
Sauvageau G, Iscove NN, Humphries RK . In vitro and in vivo expansion of hematopoietic stem cells. Oncogene 2004; 23: 7223–7232.
Brun AC, Bjornsson JM, Magnusson M, Larsson N, Leveen P, Ehinger M et al. Hoxb4-deficient mice undergo normal hematopoietic development but exhibit a mild proliferation defect in hematopoietic stem cells. Blood 2004; 103: 4126–4133.
Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T . Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J 2004; 23: 4116–4125.
Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 2004; 431: 1002–1007.
Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000; 287: 1804–1808.
Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T . In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol 2004; 6: 436–442.
Yu H, Yuan Y, Shen H, Cheng T . Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood 2006; 107: 1200–1206.
Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S et al. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med 2005; 202: 169–179.
Chung YJ, Park BB, Kang YJ, Kim TM, Eaves CJ, Oh IH . Unique effects of Stat3 on the early phase of hematopoietic stem cell regeneration. Blood 2006; 108: 1208–1215.
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 2004; 21: 843–851.
Ohta H, Sawada A, Kim JY, Tokimasa S, Nishiguchi S, Humphries RK et al. Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. J Exp Med 2002; 195: 759–770.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003; 423: 302–305.
Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol 2006; 7: 1037–1047.
Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med 2004; 199: 221–229.
Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C . Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol 2006; 7: 1048–1056.
Acknowledgements
This work was supported by NIH grant DK57899 and by the Brian Rooney Fund of the Lymphoma Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by R Knight
Rights and permissions
About this article
Cite this article
Huang, X., Cho, S. & Spangrude, G. Hematopoietic stem cells: generation and self-renewal. Cell Death Differ 14, 1851–1859 (2007). https://doi.org/10.1038/sj.cdd.4402225
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402225
Keywords
This article is cited by
-
Regulatory effect of chemokines in bone marrow niche
Cell and Tissue Research (2015)
-
Ex vivo expansion of hematopoietic stem cells
Science China Life Sciences (2015)
-
Studying the enucleation process, DNA breakdown and telomerase activity of the K562 cell lines during erythroid differentiation in vitro
In Vitro Cellular & Developmental Biology - Animal (2013)
-
Stem cells for reprogramming: could hUMSCs be a better choice?
Cytotechnology (2013)
-
Treatment with chicken-egg-white or whole-egg extracts maintains and enhances the survival and differentiation of spleen cells
Cytotechnology (2012)
