
Abstract
Elongin A is a transcription elongation factor that increases the overall rate of mRNA chain elongation by RNA polymerase II. To gain more insight into the physiological functions of Elongin A, we generated Elongin A-deficient mice. Elongin A homozygous mutant (Elongin A−/−) embryos demonstrated a severely retarded development and died at between days 10.5 and 12.5 of gestation, most likely due to extensive apoptosis. Moreover, mouse embryonic fibroblasts (MEFs) derived from Elongin A−/− embryos exhibited not only increased apoptosis but also senescence-like growth defects accompanied by the activation of p38 MAPK and p53. Knockdown of Elongin A in MEFs by RNA interference also dramatically induced the senescent phenotype. A study using inhibitors of p38 MAPK and p53 and the generation of Elongin A-deficient mice with p53-null background suggests that both the p38 MAPK and p53 pathways are responsible for the induction of senescence-like phenotypes, whereas additional signaling pathways appear to be involved in the mediation of apoptosis in Elongin A−/− cells. Taken together, our results suggest that Elongin A is required for the transcription of genes essential for early embryonic development and downregulation of its activity is tightly associated with cellular senescence.
Similar content being viewed by others


Main
Eukaryotic mRNA synthesis by RNA polymerase II (pol II) is regulated by the concerted action of a set of transcription factors that control the activity of pol II during the initiation and elongation stages of transcription. At least six general transcription factors have been identified in eukaryotic cells and found to promote the selective binding of pol II to promoters and to support the basal level of transcription.1 In addition, a diverse collection of elongation factors that promote efficient elongation of transcripts by pol II in vitro have also been identified.2, 3, 4 These factors fall into two broad functional classes based on their ability to either reactivate arrested pol II or suppress the transient pausing of pol II. The first class is composed of members of the SII family.3, 5 The second class comprises a collection of elongation factors, including TFIIF,6 Elongin,7, 8 ELL9 and CSB,10 which increase the overall rate of mRNA chain elongation by decreasing the frequency and/or duration of transient pausing of pol II at sites along the DNA template.
Elongin was identified as a heterotrimer composed of A, B and C subunits of ∼770, 118 and 112 amino acids, respectively.7, 8, 11, 12 Elongin A is the transcriptionally active subunit, whereas Elongins B and C are positive regulatory subunits that can form an isolable Elongin BC subcomplex.8, 13, 14 Although the functions of Elongin A in vivo remain largely unclear, Gerber et al.15 have recently reported that the Drosophila homolog of Elongin A (dEloA) colocalizes with pol II at sites of active transcription on polytene chromosomes, and it also plays a critical role in heat shock gene expression in vivo.15, 16 Moreover, by the RNA interference (RNAi) knockdown of dEloA, they have also shown it to be an essential factor required for proper metamorphosis, although the relationship between the elongation activity and viability remains unclear.
To gain more insight into the physiological functions of Elongin A, we have generated Elongin A-deficient mice. Elongin A−/− embryos exhibited severe growth retardation and died at between days 10.5 and 12.5 of gestation, most likely owing to excessive apoptosis. A cDNA microarray analysis of E10.5 embryos demonstrated an increased expression of hypoxia-response genes, including known transcriptional targets of p53, such as Ddit4 and Ndrg1, in Elongin A−/− embryos. Moreover, mouse embryonic fibroblasts (MEFs) derived from Elongin A−/− embryos displayed not only increased apoptosis but also the features of premature senescence, accompanied by the activation of p38 MAPK and p53. RNAi-mediated knockdown of Elongin A in MEFs also dramatically induced the senescent phenotype. A pharmacological study using SB203580 and pifithrin-α and generation of Elongin A−/− mice in p53-null background suggested that both the p38 MAPK and p53 pathways are responsible for the induction of senescence-like phenotypes, whereas additional signaling pathways, yet to be identified, appear to be involved in the mediation of apoptosis in Elongin A−/− cells. Taken together, our results demonstrate that Elongin A is an essential factor for viability, whereas also suggesting that it may regulate the transcription of genes essential for early embryonic development and that downregulation of its activity is strongly associated with cellular senescence.
Results
Elongin A−/− embryos die during embryogenesis
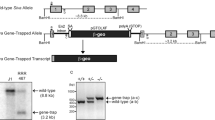
The Elongin A mutant mice were generated by homologous recombination in CCE embryonic stem (ES) cells.17 Exons 8–10 of the Elongin A gene were replaced with a neomycin cassette in the gene-targeting vector (Figure 1a).18 The mutant ES cell clones with a disrupted Elongin A locus were used to obtain the germline chimeras that, in turn, were used to generate the heterozygous F1 mutant mice. The genotypes were determined using the polymerase chain reaction (PCR), and the loss of Elongin A expression was confirmed by reverse transcriptase (RT)-PCR (Figure 1b and c). The F1 Elongin A+/− mice appeared to be normal. An analysis of live-born progeny obtained from crosses between F1 Elongin A+/− mice revealed the Elongin A mutation to be recessive embryonic lethal. To determine the timing of the death, the Elongin A genotypes the embryos at different stages of gestation were analyzed. As summarized in Table 1, Elongin A−/− embryos at E10.5 were found at a frequency that roughly matches the Mendelian distribution, thus indicating the lethality at the middle stage of embryonic development.

Generation of Elongin A-deficient mice. (a) A diagram showing the genomic organization of the Elongin A gene, the targeting vector and the targeted allele after homologous recombination. The coding exons, depicted by closed boxes, are numbered 1–11. A1, A2 and A3 or B1 and B2, indicate the primers used for PCR genotyping or RT-PCR analysis, respectively. TK, thymidine kinase cassette; NEO, neomycin resistance cassette. (b) A representative PCR-based genotype analysis of embryos from Elongin A+/− intercrosses. PCR products from the WT and mutant alleles are indicated. (c) RT-PCR analysis of Elongin A (upper panel) or the GAPDH gene (lower panel) in Elongin A+/+, Elongin A+/−, and Elongin A−/− MEFs. (d) Expression of Elongin A in mouse embryos. Total RNA, isolated from embryos at various indicated developmental stages, was subjected to a Northern blot analysis with cDNA probes
In a previous study in adult mice, Elongin A mRNA was detected in all tissues examined.19 To examine whether Elongin A is expressed when such lethality occurs, we analyzed its expression at different developmental stages in wild-type (WT) embryos. A high level of Elongin A mRNA was detected in E4.5 embryos and was thereafter sustained throughout embryogenesis (Figure 1d), suggesting that Elongin A plays a critical role in embryonic development.
Elongin A−/− embryos exhibit growth retardation and increased apoptosis
Gross inspection and fine histological examinations of live E10.5 Elongin A−/− embryos revealed a wide variety of severe developmental abnormalities. Overall, Elongin A−/− embryos were consistently smaller, and also severely more hypoplastic, than controls (Figure 2a–e). More specifically, Elongin A−/− embryos exhibited cardiac hypoplasia with abnormally thin ventricular walls, showing a minimum thickness of one cell layer, and dilated ventricles that presumably arose from the effects secondary to severe heart failure (Figure 2f and g). A poor development of the central nervous system and liver was also prominent. The thickness and cellularity of the spinal and cranial neuroepithelium were significantly reduced in Elongin A−/− embryos (Figure 2h and i). The livers of Elongin A−/− embryos were markedly decreased in size and composed of irregular trabeculae with fewer hepatocytes than in WT embryonic livers (Figure 2j, k). In addition to these defects, Elongin A−/− embryos were strikingly paler than the controls, but the presence of some nucleated red blood cells suggested that yolk sac hematopoiesis was only partially blocked (Figure 2a–c and data not shown).

Morphology and histology of E10.5 embryos with different Elongin A genotypes. (a–c) Gross morphology of Elongin A+/+ (a), Elongin A+/− (b), and Elongin A−/− (c) embryos. Note the small size and paleness of Elongin A−/− embryos. (d, e) Overview of sagittal sections of Elongin A+/+ (d) and Elongin A−/− (e) embryos. Scale bars, 1 mm. (f, g) Heart sections from Elongin A+/+ (f) and Elongin A−/− (g) embryos. Magnification, × 100. Note the cardiac hypoplasia with significantly thinner ventricular walls (arrow) in the Elongin A−/− embryo. (h, i) Forebrain sections from Elongin A+/+ (h) and Elongin A−/− (i) embryos. Magnification, × 100. Note the reduced thickness and cellularity of the cranial neuroepithelium in the Elongin A−/− embryo (arrow). (j, k) Liver sections from Elongin A+/+ (j) and Elongin A−/− (k) embryos. Magnification, × 200. Note the smaller liver and irregular trabecular structure in the Elongin A−/− embryo (arrow). (l, m) A TUNEL analysis of sagittal sections from Elongin A+/+ (l) and Elongin A−/− (m) embryos. Magnification, × 400. Large numbers of apoptotic cells are detected in the Elongin A−/− embryo. (n–s) Sagittal sections of Elongin A+/+ (n, o, p) and Elongin A−/− (q, r, s) embryos at E10.5 were stained with anti-Ki67 antibody to visualize proliferating cells (n, q), with DAPI to visualize the nuclei (o, r), and with both anti-Ki67 and DAPI (p, s). Scale bar, 10 μm. (t, u) Incorporation of BrdU in Elongin A+/+ (t) and Elongin A−/− (u) embryos at E10.5. Scale bars, 10 μm
To clarify the cause of the developmental growth defect of Elongin A−/− embryos, we carried out both apoptosis and cell cycle analyses. A terminal dUTP nick end labeling (TUNEL) assay on E10.5 embryo sections revealed that massive numbers of TUNEL-positive cells were readily detectable in Elongin A−/− embryos, whereas very few cells were positive in WT embryos (Figure 2l, m). For the cell cycle analysis, we examined the incorporation of 5-bromo-2′-deoxyuridine (BrdU) and the expression of Ki67 nuclear protein in E10.5 embryos. The proportions of cells positive for each of these proliferation markers were similar in WT and Elongin A−/− embryos (Figure 2n–u), suggesting that the rate of cell proliferation in vivo did not significantly decrease owing to the loss of Elongin A.
Elongin A−/− MEFs display apoptosis and senescence-like growth defect
To rule out the possibility that the growth defect of Elongin A−/− embryos was due to extra-embryonic abnormalities, we examined the growth potential and apoptotic status of Elongin A−/− MEFs in vitro. Similar to the embryonic phenotype, MEFs prepared from Elongin A−/− embryos exhibited severe growth defects and a significant increase in the apoptotic population (Figure 3a, b). Therefore, these in vivo and in vitro data suggest that Elongin A−/− embryos have intrinsic growth defects and therefore undergo net cell loss, at least in part, owing to apoptotic cell death.

Increased apoptosis and senescence-like growth defect in Elongin A−/− MEFs. (a) Growth curves for Elongin A+/+ Elongin A+/−, and Elongin A−/− MEF cultures at passage 2. The cultures were harvested at the indicated time points, and the total number of cells was determined and normalized to the number of cells at day 1 (24 h after plating). The average for three independently derived MEF cultures of each genotype analyzed in duplicate is shown. (b) An analysis of apoptosis in Elongin A+/+, Elongin A+/− and Elongin A−/− MEFs. The population of apoptotic cells was determined by annexin V-FITC and PI staining. Typical annexin V/PI FACS profiles of MEFs are shown. The percentage of cells in each fraction is shown below. (c) An analysis of the percentage of actively cycling cells in Elongin A+/− and Elongin A−/− MEFs. MEFs at passage 2 were continuously labeled with BrdU for 48 h and then stained with anti-BrdU to detect DNA synthesis (vertical axis) and with PI to detect total DNA (horizontal axis). The numerical data are the averages of measurements with three independently derived MEF cultures of each genotype. FACS profiles for a typical experiment are shown as an example. (d) Morphology of MEFs at passage 2. Elongin A−/− MEFs showed a flattened and enlarged morphology compared with the Elongin A+/+ MEFs. (e) SA-β-gal staining of MEFs at passage 2 (left panel) and percentages of cells positive for this staining (right panel). Elongin A−/− MEFs showed an increase in the number of SA-β-gal-positive cells. *P>0.05. **P<0.05
Next, to test whether cell cycle regulation is affected in Elongin A−/− MEFs, BrdU incorporation assays were performed. In contrast to the findings in vivo, a moderate increase in the G0/G1 phase population accompanied by a concomitant decrease in the S-phase population was observed in Elongin A−/− MEFs (Figure 3c), suggesting that the number of cells actively cycling was decreased in Elongin A−/− MEFs. We also found that many Elongin A−/− MEFs displayed flattening and cytoplasmic enlargement, morphological features consistent with senescent cells, as early as after the first passage of culture (Figure 3d). We therefore checked the activity of senescence-associated β-galactosidase (SA-β-gal), a biochemical marker of senescence,20 in these cells. As shown in Figure 3e, approximately 30% of Elongin A−/− MEFs accumulated SA-β-gal, whereas very few of the controls displayed this marker, suggesting that the growth inhibition observed in Elongin A−/− MEFs was partly due to cellular senescence.
Expression of hypoxia-response genes is increased in Elongin A−/− embryos
To investigate whether the lack of Elongin A affects the expression levels of total mRNA, a dot blot analysis was performed. As shown in Figure 4a, the total mRNA levels in Elongin A−/− embryos were equivalent to those in the control embryos.

Increased expression of hypoxia-responsive genes in Elongin A−/− embryos at E10.5. (a) Comparison of total mRNA levels in Elongin A+/+, Elongin A+/− and Elongin A−/− embryos at E10.5. (b) Identification of genes differentially expressed in Elongin A+/+ and Elongin A−/− whole embryos using a microarray analysis. Genes upregulated more than twofold in Elongin A−/− embryos are listed (left panel) and the results were confirmed by RT-PCR assays (right panel). The asterisks indicate hypoxia-response genes. (c) A Western blot analysis of HIF-1α and HIF-2α from lysates of Elongin A+/+, Elongin A+/− and Elongin A−/− whole embryos. α-Tubulin was used as a loading control
Next, to examine the effects of Elongin A deletion on the expression levels of individual mRNAs, a cDNA microarray analysis comparing WT and Elongin A−/− embryos was performed. As shown in Figure 4b, of all genes analyzed only 0.4% showed increases of expression levels of more than twofold and only 0.3% showed decreases of more than twofold owing to the lack of Elongin A. A differential expression of some of these genes was confirmed by semiquantitative RT-PCR. Most notably, a considerable proportion of the genes whose expression increased markedly in Elongin A−/− embryos were those known to be upregulated under hypoxic conditions. To investigate the mechanism of the induction of hypoxia-responsive genes in Elongin A−/− embryos, the expression level of hypoxia-inducible factor-1 (HIF-1) was analyzed. HIF-1 is a heterodimer that consists of the hypoxic response factor HIF-1α and the constitutively expressed HIF-1β.21, 22 We therefore checked the expression of HIF-1α and its homolog HIF-2α by Western blotting. As shown in Figure 4c, however, the protein levels of both HIF-1α and HIF-2α were not increased in the Elongin A−/− embryos, suggesting the possibility that other pathways, independent of HIF-1, may also play some role in the observed upregulation of the hypoxia-response genes.
Activation of p53 and p38 MAPK in Elongin A−/− embryos and MEFs
Those genes upregulated in the Elongin A−/− embryos, such as Ddit4 and Ndrg1,23, 24 are also known to be direct transcriptional targets of p53, and their translational products are demonstrated to play an important role in p53-dependent apoptosis. In addition, hypoxia and the blockage of transcription elongation by agents such as UV light, camptothecin or actinomycin D have been shown to induce the phosphorylation of human p53 at Serine 15, resulting in the stabilization of the protein.25, 26 We therefore checked whether this modification also occurs in Elongin A−/− embryos. As shown in Figure 5a, Elongin A−/− embryos indeed showed an increased phosphorylation of p53 at Ser18 (corresponding to Ser15 in human p53) in comparison to those of controls, thus suggesting that the p53 signaling pathway is also activated in Elongin A−/− embryos.

The activation of p53 and p38 MAPK in Elongin A−/− embryos and MEFs. (a) Western blot analysis of p53 phosphorylation at Ser18, from lysates of Elongin A+/+, Elongin A+/− and Elongin A−/− whole embryos. α-Tubulin was used as a loading control. (b) An expression analysis of various cell cycle regulatory proteins. Elongin A+/+, Elongin A+/− and Elongin A−/− MEFs at passage 2 were analyzed by Western blotting using the indicated antibody; α-tubulin was used as a loading control. (c) Western blot analysis of p38 MAPK (p38, total p38; and P-p38, phosphorylated p38) from lysates of Elongin A+/+, Elongin A+/− and Elongin A−/− MEFs at passage 2 (left panel) and whole embryos (right panel); α-tubulin was used as a loading control
Cellular senescence has been shown to be typically accompanied by the activation of p53 and pRb, along with an increased expression of cyclin-dependent kinase inhibitors, such as p16INK4a, p19ARF and p21WAF1.27 To characterize the nature of the senescence-like growth defect of Elongin A−/− MEFs, we examined the expression of several key regulators of the cell cycle (Figure 5b). The expression levels of p16INK4a, p19ARF and cyclin D1 were dramatically increased in Elongin A−/− MEFs. The induced expression of cyclin D1 is one of the characteristic features observed in senescent cells;28 therefore, the accumulation of cyclin D1 may have a rather inhibitory effect on cell growth. Modest increases in the levels of p53 and p21WAF1 and a significant decrease in hyperphosphorylated pRb, were also observed in Elongin A−/− MEFs.
Although the precise molecular mechanisms remain unclear, the activation of the p38 MAPK pathway has been reported to contribute to the induction of cellular senescence.29, 30 To explore the possible involvement of p38 MAPK in the induction of senescence-like phenotypes in Elongin A−/− MEFs, we examined the levels of total and activated p38 MAPK in passage to MEFs and E10.5 whole embryos. The total amounts of p38 MAPK were essentially the same; however, a significant increase in the amount of phosphorylated p38 MAPK was observed in Elongin A−/− MEFs (Figure 5c, left panel). Contrary to the results in MEFs, however, no increase in the phosphorylated p38 MAPK was observed in the Elongin A−/− embryos (Figure 5c, right panel), thus suggesting that the onset of the senescence process in Elongin A−/− cells occurred during the ex vivo culture of MEFs, and not in utero.
Senescence-like phenotype, but not apoptosis, in Elongin A−/− MEFs is rescued by the inhibition of both p53 and p38 MAPK
To investigate whether the activated p53 and p38 MAPK really contribute to the induction of apoptosis and senescence-like phenotypes in Elongin A−/− MEFs, we cultured Elongin A−/− MEFs either with or without SB203580, which specifically inhibits the kinase activities of the α and β isoforms of p38 MAPK,31 and pifithrin-α, which specifically inhibits p53.32 We first examined the effect of these inhibitors on cell growth. Elongin A−/− MEFs treated with SB203580 showed a partial recovery, whereas those treated with either pifithrin-α or both inhibitors showed a complete recovery of cell growth (Figure 6a). We next examined the effect of these inhibitors on apoptosis. In contrast, the SB203580 treatment resulted in a partial reduction in the percentage of apoptotic cells in Elongin A−/− MEFs, but treatment with neither pifithrin-α nor both inhibitors had a significant effect on the apoptotic population (Figure 6b). We next examined the effect on the cell cycle status using BrdU incorporation assays. Elongin A−/− MEFs treated with SB203580 showed a moderate but significant increase in the proportion of actively cycling cells, whereas those treated with either pifithrin-α or both inhibitors demonstrated a complete recovery to a level even higher than that of untreated Elongin A+/− MEFs (Figure 6c). Finally, we examined the effect on the SA-β-gal activity. Treatment of Elongin A−/− MEFs with SB203580 resulted in a partial reduction of the percentage of cells positive for SA-β-gal activity. Combined treatment with both SB203580 and pifithrin-α resulted in complete inhibition, whereas that with pifithrin-α alone had no significant effect (Figure 6d). These results indicate that the activation of both p53 and p38 MAPK is responsible for mediating senescence-like growth defects, but not apoptosis, in Elongin A−/− MEFs.

Effects of p53 and p38 MAPK inhibitors on apoptosis and senescence-like growth defect in Elongin A−/− MEFs. (a) Representative growth curves documenting the effects of pifithrin-α (PFT) and SB203580 (SB) on the growth of Elongin A+/− and Elongin A−/− MEFs at passage 2. (b) Effects of PFT and SB on the percentage of apoptotic cells in Elongin A+/− and Elongin A−/− MEFs. *P<0.05. (c) Effects of PFT and SB on the percentage of actively cycling cells in Elongin A+/− and Elongin A−/− MEFs. *P<0.05. **P<0.005. (d) Effects of PFT and SB on the SA-β-gal staining (upper panel) and percentages of cells positive for this staining (lower panel) in Elongin A+/− and Elongin A−/− MEFs at passage 2. *P<0.05. **P<0.005
p53 mutation fails to rescue apoptosis and embryonic lethality in Elongin A−/− mice
In order to analyze the importance of p53 activation in apoptosis and lethality observed in Elongin A−/− embryos, we first generated Elongin A+/− p53+/− mice by crossing Elongin A+/− and p53+/− mice. We then interbred these mice and analyzed the genotypes of live-born progeny. Of the 117 mice analyzed, however, none were genotyped as Elongin A−/− p53−/− (Table 2). This indicates that the loss of p53 does not rescue the embryonic lethality caused by the Elongin A−/− mutation. Next, to assess the effect of p53 on apoptosis, we carried out cell death detection ELISA assays of E10.5 Elongin A−/− p53+/+ and Elongin A−/− p53−/− embryos. Consistent with the findings obtained with the TUNEL assays (Figure 2l, m), Elongin A−/− embryos showed significantly increased apoptosis compared with the WT embryos (Figure 7a). However, the extent of apoptosis in Elongin A−/− embryos was not decreased in the p53−/− background (Figure 7b). In addition, Elongin A−/− p53+/+ and Elongin A−/− p53−/− embryos at E10.5 appeared to be morphologically undistinguishable (data not shown). These results indicate that a loss of p53 is not sufficient to overcome the apoptosis triggered by a deficiency of Elongin A.

Effect of p53 mutation on the level of apoptosis in Elongin A−/− embryos. (a) A quantitative analysis of the cytoplasmic histone-associated DNA fragments in Elongin A+/+ and Elongin A−/− embryos at E10.5. (b) Effect of p53 mutation on the level of apoptosis in Elongin A+/+ and Elongin A−/− embryos at E10.5. The level of apoptosis in embryos was determined by ELISA quantification of cytoplasmic histone-associated DNA fragments
Induction of cellular senescence by RNAi-mediated knockdown of Elongin A
To confirm the role of Elongin A in cellular senescence and apoptosis, we knocked down Elongin A in WT MEFs by RNAi-mediated gene silencing. Two independent sequences of Elongin A were used as targets of RNAi. As shown in Figure 8a, MEFs transfected with plasmid vectors encoding either EloA1-1 or EloA1-2 short hairpin RNA (shRNA) specific for Elongin A mRNA exhibited a significant reduction in Elongin A mRNA levels compared with that apparent in MEFs transfected with a control vector. Expression of Elongin A shRNAs did not affect the expression of 2′5′-oligoadenylate synthetase (OAS1), a classic interferon target gene that is induced >50-fold on activation of an interferon response.33 MEFs expressing Elongin A shRNAs exhibited a phenotypic change similar to those observed in MEFs derived from Elongin A−/− embryos, such as flattening, cytoplasmic enlargement and increase in SA-β-gal staining (Figure 8b, c), accompanied by increase in the amount of phosphorylated p38 MAPK, and increased p16INK4a, p19ARF, p53, p21WAF1 and cyclin D1 expression (Figure 8d). Contrary to Elongin A−/− mice, however, neither increased p53 Ser18 phosphorylation, activation of the hypoxia-response genes, nor induction of apoptosis was observed in MEFs expressing Elongin A shRNAs (Figure 8d–f). These results indicate that cellular senescence, but not apoptosis, can be induced in MEFs by RNAi-mediated knockdown of Elongin A.

Induction of cellular senescence in MEFs by RNAi knockdown of Elongin A. (a) RT-PCR analysis of Elongin A (upper panel), OAS1 (middle panel) or the GAPDH gene (lower panel) in MEFs 7 days after the transfection of control or Elongin A shRNA (EloA1-1 or EloA1-2) vector. (b) Morphology of MEFs transfected with control or Elongin A shRNA vector at day 7. (c) SA-β-gal staining of MEFs transfected with control or Elongin A shRNA vector at day 7 (upper panel), and percentages of cells positive for this staining (lower panel). (d) Phosphorylation status of p38 MAPK and p53 Ser18, and expression of various cell cycle regulatory proteins were analyzed by Western blotting using lysates of MEFs transfected with control or Elongin A shRNA vector at day 7. α-Tubulin was used as a loading control. (e) RT-PCR analysis of various hypoxia-response genes in MEFs transfected with control or Elongin A shRNA vector at day 7. (f) A quantitative analysis of the cytoplasmic histone-associated DNA fragments in MEFs transfected with control or Elongin A shRNA vector at day 5
Discussion
Numerous in vitro studies have demonstrated a requirement for the transcription elongation factors in transcriptional regulation; however, the functions of these factors in vivo have not been satisfactorily clarified.2, 3, 4 We therefore generated mice lacking one of these factors, Elongin A. Elongin A−/− embryos were severely retarded in development and died between days 10.5 and 12.5 of gestation, most likely owing to extensive apoptosis. Moreover, MEFs derived from Elongin A−/− embryos exhibited not only increased apoptosis but also senescence-like growth defects, accompanied by the activation of p38 MAPK and p53. Knockdown of Elongin A in MEFs by RNAi also dramatically induced the senescent phenotype.
The expression of hypoxia-response genes was strongly increased in the Elongin A−/− embryos at E10.5 in comparison with the controls. What might be the mechanism for such upregulation? It is possible that an activated expression of the hypoxia-response genes as a physiological adaptation to hypoxia could terminate in WT embryos as the development of the cardiovascular system reaches a level that would allow the supply of sufficient oxygen to all tissues at this stage.21 On the other hand, it might continue in Elongin A−/− embryos because a developmental growth defect owing to Elongin A deficiency leads to hypoplasia of the heart, which thus causes systemic hypoxia. Furthermore, the induction of the hypoxia-response gene expression is thought to be primarily mediated through the hypoxia-responsive transcription factor HIF-1;21, 22 however, the protein levels of neither HIF-1α nor HIF-2α, two key regulatory components of HIF-1, were observed to be increased in the Elongin A−/− embryos in comparison to the controls. It is noteworthy that p53 inhibits the hypoxia-induced expression of HIF-1α by promoting its MDM2-mediated ubiquitination and proteasomal degradation.34 It therefore seems possible that other pathways independent of HIF-1 may also play some roles in the activation of hypoxia-response genes in Elongin A−/− embryos.22, 35, 36 In any case, the identification of the genes whose transcription is regulated by the action of Elongin A is thus considered to be essential to fully understand the mechanism of the activation for hypoxia-response genes in Elongin A−/− embryos.
We have examined whether inhibitors of p53 and/or p38 MAPK are capable of rescuing the phenotypes of Elongin A−/− MEFs. Pifithrin-α rescued the growth defects of Elongin A−/− MEFs without restoring the flattened and enlarged morphology. On the other hand, SB203580 restored the morphology of Elongin A−/− MEFs to a level nearly equal to that of the control MEFs, even though it only partially rescued the growth defects. These findings suggest that, although p53 is essential for growth defects, p38 MAPK plays a more dominant role in the senescence-associated changes of cell morphology in Elongin A−/− MEFs (Figure 9). Furthermore, as cellular senescence is irreversible, the finding that pretreatment with both SB203580 and pifithrin-α can rescue senescence-like phenotypes suggests that the senescence process in Elongin A−/− cells occurs during the ex vivo culture of MEFs, not in utero. Consistent with this assumption, neither the amount of phosphorylated p38 MAPK nor the proportion of cells positive for proliferation markers was significantly altered in the Elongin A−/− embryos in comparison to the controls at E10.5.

A working model summarizing how Elongin A inactivation results in cellular senescence, apoptosis and embryonic lethality
In contrast to the senescence-like phenotypes, the inhibition of p53 neither by pifithrin-α nor by p53-null background rescued the accelerated apoptosis in Elongin A−/− MEFs or embryos, respectively. As activation of hypoxia-response genes was observed in Elongin A−/− embryos and both the p53-dependent and independent pathways have been demonstrated to be involved in the signaling of the hypoxic response,22, 37, 38 we assume that the hypoxic signaling pathway independent of p53 may thus play a more important causative role in the induction of apoptosis in Elongin A−/− mice (Figure 9). Consistent with our assumption, increased apoptosis was not observed in MEFs expressing Elongin A shRNAs, which were shown to be free of the upregulation of hypoxia-response genes.
Finally, an expression analysis of mRNA demonstrated the total mRNA levels in Elongin A−/− embryos to be equivalent to those in the control embryos and that the expression of only 0.3% of all genes analyzed were decreased more than twofold in the Elongin A−/− embryos in comparison with the WT embryos, suggesting that Elongin A regulates the transcription of only a small subset of genes. As Elongin A−/− mice showed lethality at the middle stage of embryonic development, Elongin A might be predicted to play a crucial role for the expression of at least one of those genes, whose expression must be precisely timed during development. The identification and functional analysis of Elongin A-responsive genes will help us to better understand how the loss of Elongin A contributes to embryonic lethality and the induction of apoptosis and cellular senescence.
Materials and Methods
Generation of Elongin A-deficient mice
Elongin A mutant ES cell clones were generated as described previously 18 and microinjected into blastocysts for the generation of chimeric mice. Chimeric males were crossed with C57BL/6 females, and agouti-colored offspring were analyzed for the germline transmission of the Elongin A mutation. Heterozygous animals were intercrossed to generate homozygous mutant animals.
Generation of Elongin A−/− p53−/− mice
Mice mutant for p53 were obtained from RIKEN BRC.39 Elongin A+/− mice were bred to p53 mutant mice, and the genotypes of the progeny were then determined by a PCR analysis.
PCR genotyping
Genomic DNA was isolated from mouse-tail biopsy samples and embryonic yolk sacs, and genotyping was performed using PCR. For Elongin A, a mixture of primers was used to detect WT and mutant alleles: A1, 5′-TCCTGAGGAGCTGGCCTATG-3′ (wild-type); A2, 5′-ACCGCCTGTGCTCTAGTAGC-3′ (mutant allele); and A3, 5′-TTATCGCCGGGAGAATCTG-3′ (common). The conditions for PCR were 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, for 30 cycles. The amplified PCR products of a 700-base pair (bp) fragment and a 600-bp fragment from WT and mutant alleles, respectively, were resolved on 2% agarose gels and stained with ethidium bromide. For p53, a set of primers, LCB614 5′-GTTATGCATCCATACAGTACA-3′ and LCB651 5′-CAGGATATCTTCTGGAGGAAG-3′, was used. The conditions were 94°C for 30 s, 55°C for 30 s and 72°C for 90 s, for 35 cycles. The products of a 400-bp fragment and a 1500-bp fragment from WT and mutant alleles, respectively, were resolved on 1% agarose gel and then were stained with ethidium bromide.
For the paraffin sections of embryos, DNA was isolated using DEXPAT (TaKaRa) according to the manufacturer's instructions, precipitated with ethanol and dissolved in 0.1 × Tris–EDTA (TE) buffer. Genotyping was performed using nested PCR. The primers for the first PCR were: A1 (WT allele); C1, 5′-AGTAGCCGTTATTAGTGGAGAGG-3′ (mutant allele); and C2, 5′-CAGCAACAGGTCTCACCATGAG-3′ (common). The primers for the nested PCR were: C3, 5′-CCAGTACTAGCAGTGCCCAC-3′ (WT allele); A2 (mutant allele); and C4, 5′-GCCAGCCTGTCCTTGGCTCAG-3′ (common). The conditions for the first PCR were 94°C for 20 s, 61°C for 20 s and 72°C for 20 s, for 35 cycles. The product of the first PCR was diluted 1:50 in 0.1 × TE buffer, and 2 μl was used for the nested PCR. The conditions for the nested PCR were 94°C for 20 s, 60°C for 20 s and 72°C for 20 s, for 35 cycles. The amplified PCR products of a 160-bp fragment and a 120-bp fragment from WT and mutant alleles, respectively, were resolved on 3% agarose gel and then were visualized with UV light after staining with ethidium bromide.
RT-PCR analysis
Total RNA was isolated from either whole embryos or MEFs using an RNeasy Mini kit (Qiagen) according to the manufacturer's protocol. First-strand cDNA was synthesized from 1 μg of total RNA, with an oligo-dT primer and MuLV reverse transcriptase (Applied Biosystems). One-twentieth of each RT reaction product was used for PCR amplification. The primer sets used are B1 (5′-TGGCCCAGTGGAAGAAGCTGGTTCCAG-3′) and B2 (5′-GGCCTTTCTTCCTTAAAGTCTCGGTGACAGTG-3′) for Elongin A, TRIB3-F (5′-GTTTCCTGCAGGAAGAAACCG-3′) and TRIB3-R (5′-GTGCCACGGCACTAGCCATC-3′) for Tribbles 3 (Trib3), STC2-F (5′-CAGTTTGTGACCCTGGCTTTG-3′) and STC2-R (5′-CACAATCACACCGACGTTCTC-3′) for Stanniocalcin 2 (Stc2), DDIT4-F (5′-CTTCTGTGCGCCTTCATTC-3′) and DDIT4-R (5′-CTCAGGTGGCTATCGTCAGT-3′) for DNA-damage-inducible transcript 4 (Ddit4), NDRG1-F (5′-ACAACCCCCTCTTCAACTCTG-3′) and NDRG1-R (5′-CATTCCACCACAGCATCCACG-3′) for N-myc down-regulated gene 1 (Ndrg1), NIP3-F (5′-CTGGGTAGAACTGCACTTCAG-3′) and NIP3-R (5′-GCGCTTCGGGTGTTTAAAAAGG-3′) for Bcl-2-interacting protein 1 (Bnip3), HIG1-F (5′-GTCAACCAACACAGACCTTTCTC-3′) and HIG1-R (5′-CTAAGGCTTAGGCTTAGGGTTG-3′) for Hypoxia-induced gene 1 (Hig1), VEGFa-F (5′-CTTGGGTGCACTGGACCCTG-3′) and VEGFa-R (5′-GGTTTGATCCGCATGATCTGC-3′) for vascular endothelial growth factor A (Vegfa), p21-F (5′-AATCCTGGTGATGTCCGACC-3′) and p21-R (5′-AAAGTTCCACCGTTCTCGG-3′) for p21WAF1 and OAS1-F (5′-TGTGCGAGGTTCAGCATGAG-3′) and OAS1-R (5′-ACTGTCCAGTAGATTTGAAG-3′) for OAS1, and mGA1 (5′-CTGCCATTTGCAGTGGCAAAG-3′) and mGA2 (5′-TGGTATTCAAGAGAGTAGGGA-3′) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All cDNAs were amplified in the quantitative ranges: 20 cycles for Ddit4, Bnip3 and Hig1, 25 cycles for Trib3, Stc2, Ndrg1 and Vegfa, 30 cycles for Elongin A and GAPDH, and 35 cycles for p21 and OAS1. The amplified PCR products were resolved on 1–2% agarose gel and then were visualized with UV light after staining with ethidium bromide.
Northern blot analysis
Mouse Embryo Full Stage Blot (Seegene), containing 20 μg of total RNA per sample, was hybridized using ExpressHyb solution (Clontech) under stringent conditions as recommended by the manufacturer. The mouse Elongin A probe contained the sequence encoding amino acids 97–468, and the blot was stripped and reprobed using GAPDH cDNA.
Dot blot analysis
To determine the total mRNA level in the E10.5 embryos with different Elongin A genotypes, 1 μg of heat-denatured RNA (70°C for 3 min) was added to 100 ml of 1 M ammonium acetate and blotted onto a nylon filter (Hybond-N+, Amersham Biosciences) with a dot blotter manifold (Sanplatec). The filter was air-dried and prehybridized overnight at 37°C in 4 × SSC (1 × SSC is 0.15 M NaCl, 0.015 M Na-citrate), 0.2% Ficoll, 0.2% polyvinylpyrrolidone, 0.5% sodium dodecyl sulfate (SDS) and 200 mg/ml denatured salmon sperm DNA, followed by hybridization at 37°C with 8 mCi/ml of 32P-labeled poly(dT) in the prehybridization buffer for 12–20 h. The filter was washed at 37°C for 15 min with the prehybridization buffer and then three times at room temperature for 20 min each time in 1 × SSC and 0.1% SDS.
Microarray analysis
Total RNA was isolated from Elongin A+/+ and Elongin A−/− E10.5 embryos according to the manufacturer's protocol using the RNeasy Mini kit (Qiagen). Aliquots (1 μg) of total RNA were labeled using the Agilent Linear Amplification/Labeling kit (Agilent Technologies) according to the manufacturer's instructions. After checking the labeling efficiency, 1 μg aliquots of Cy3-labeled Elongin A+/+ cRNA and Cy5-labeled Elongin A−/− cRNA were mixed, then hybridized to an Agilent Whole Mouse Oligo Microarray using the manufacturer's hybridization protocol. After the washing step, the microarray slide was analyzed with an Agilent Microarray scanner and software (scanner model G2505B, software G2565BA). A data analysis was performed using Agilent Feature Extraction software (Version A.6.1.1).
Histological analysis
Pregnant mice were killed at E10.5, and whole uteri were fixed in 4% paraformaldehyde for several hours, then in 20% neutral buffered formalin overnight. After fixation, the embryos were carefully dissected, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin.
A TUNEL assay was performed on paraffin sections of E10.5 embryos using the ApopTag Plus Peroxidase In situ Apoptosis Detection kit (Serologicals) according to the manufacturer's instructions.
Immunostaining with anti-Ki67 nuclear antigen (YLEM) was performed following the manufacturer's instructions. An Alexa 488-conjugated anti-rabbit antibody (Molecular Probes) was used as the secondary antibody, and slides were also stained with DAPI to visualize all nuclei.
For BrdU experiments, pregnant mice at E10.5 were injected intraperitoneally with BrdU (Sigma) in phosphate-buffered saline (PBS) at a dose of 100 μg/g of body weight. The mice were killed 2 h after the injection, and the embryos were fixed with 4% paraformaldehyde. The fixed sections were denatured by incubation in 1 N HCl at 65°C for 30 min and then were stained with anti-BrdU antibody (Roche).
Mouse embryonic fibroblast assays
MEFs were prepared from either E10.5 or E13.5 embryos. Fetal tissue samples were rinsed in PBS and then were mechanically dissociated in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). The cells were then plated out on six-well plates. These cells were considered to be passage 1 MEFs.
For growth curves, 2.5 × 104 cells were plated in triplicate into 12-well plates. At various time points, the cells were trypsinized and counted using a hemocytometer.
For the quantitative detection of apoptotic cells, both adherent and floating cells were harvested and stained with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) using the Annexin V-FITC Apoptosis Detection kit (BioVision) according to the manufacturer's instructions. The stained cells were analyzed with a FACSCalibur (Becton Dickinson) using the CELL Quest software package (BD Biosciences).
For the cell cycle analysis, MEFs grown in DMEM with 10% FBS were metabolically labeled with 10 μM BrdU for 48 h. The cells were harvested and fixed in 70% ethanol at 4°C overnight. The cells were then treated with 2 N HCl and 0.5% Triton X-100 for 30 min at room temperature, followed by neutralization with 0.1 M Na2B4O7 (pH 8.5) and subsequent incubation with FITC-conjugated anti-BrdU antibody (Progen) for 1 h at room temperature. Finally, the cells were washed with PBS and 0.5% Tween 20, resuspended in PBS containing 5 μg/ml PI, and analyzed with a FACSCalibur (Becton Dickinson) using the CELL Quest software package (BD Biosciences).
For the pharmacological inhibition of p38 MAPK and p53 activity, SB203580 (BIOMOL) and pifithrin-α (Calbiochem), respectively, were dissolved in dimethylsulfoxide (DMSO) and added to the cultures immediately after the isolation of MEFs.
Senescence-associated β-galactosidase assay
SA-β-gal activity at pH 6.0 was measured using the Senescence β-galactosidase Staining kit (Cell Signaling) according to the manufacturer's instructions.
Quantitative analysis of apoptosis in whole embryos
For the quantitative analysis and to determine the cytoplasmic histone-associated DNA fragments, which are indicative of on-going apoptosis, the Cell Death Detection ELISAPLUS (Roche Applied Science) was used according to the manufacturer's instructions.
Western blot analysis
Whole-cell extracts of E10.5 embryos or MEFs were prepared in lysis buffer (50 mM Tris-HCl (pH 7.0), 2% SDS, 10% glycerol, 50 mM NaF and 1 mM Na3VO4) containing the Complete Protease Inhibitor Cocktail (Roche Applied Science). The protein concentration was determined by using the Bradford assay, and 75 μg of each extract was fractionated by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore). The following primary antibodies were used: HIF-1α (H1α67, Novus Biologicals), HIF-2α (Novus Biologicals), Rb (C-15, Santa Cruz), p53 (FL-393 from Santa Cruz and CM5 from Novocastra), phospho-p53 (human Ser15, Cell Signaling), p16INK4a (M-156, Santa Cruz), p19ARF (Neo Markers), p21WAF1 (C-19, Santa Cruz), cyclin D1 (DCS-6, MBL), p38 (Cell Signaling), phospho-p38 (Thr180/Tyr182, Cell Signaling), and α-tubulin (DM1A, Sigma). Horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse antibodies (Amersham) were used as secondary antibodies. Detection was performed using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce).
RNA interference
DNA corresponding to an shRNA sequence was subcloned into the SacI and KpnI sites of piGENE-tRNA-Neo vector (iGENE Therapeutics), which expresses tRNA-shRNA under the control of the human tRNAVal promoter. The DNA for the shRNA encodes a 30-nucleotide hairpin sequence specific to the mRNA target, with a loop sequence (AAAACTCGAGAAAA) separating the two complementary domains. The hairpin sequences specific for mouse Elongin A mRNA correspond to nucleotides 1111 to 1140 (EloA1-1) and 2077 to 2106 (EloA1-2) of the coding region. E13.5 MEFs were transfected with these vectors using FuGENE 6 (Roche) according to the manufacturer's protocol. After 24 h, the transfected cells were selected in medium containing 400 μg/ml G418 (Sigma) for 4–6 days and were used for experiments.
Abbreviations
- HIF-1:
-
hypoxia-inducible factor-1
- MEF:
-
mouse embryonic fibroblast
- OAS1:
-
2′5′-oligoadenylate synthetase
- pol II:
-
RNA polymerase II
- RNAi:
-
RNA interference
- SA-β-gal:
-
senescence-associated β-galactosidase
- shRNA:
-
short hairpin RNA
References
Roeder RG . The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 1996; 21: 327–335.
Conaway JW, Shilatifard A, Dvir A, Conaway RC . Control of elongation by RNA polymerase II. Trends Biochem Sci 2000; 25: 375–380.
Shilatifard A, Conaway RC, Conaway JW . The RNA polymerase II elongation complex. Annu Rev Biochem 2003; 72: 693–715.
Sims III RJ, Belotserkovskaya R, Reinberg D . Elongation by RNA polymerase II: the short and long of it. Genes Dev 2004; 18: 2437–2468.
Wind M, Reines D . Transcription elongation factor SII. BioEssays 2000; 22: 327–336.
Price DH, Sluder AE, Greenleaf AL . Dynamic interaction between a Drosophila transcription factor and RNA polymerase II. Mol Cell Biol 1989; 9: 1465–1475.
Bradsher JN, Jackson KW, Conaway RC, Conaway JW . RNA polymerase II transcription factor SIII. I. Identification, purification, and properties. J Biol Chem 1993; 268: 25587–25593.
Aso T, Lane WS, Conaway JW, Conaway RC . Elongin (SIII): a multisubunit regulator of elongation by RNA polymerase II. Science 1995; 269: 1439–1443.
Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW . An RNA polymerase II elongation factor encoded by the human ELL gene. Science 1996; 271: 1873–1876.
Selby CP, Sancar A . Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci USA 1997; 94: 11205–11209.
Garrett KP, Tan S, Bradsher JN, Lane WS, Conaway JW, Conaway RC . Molecular cloning of an essential subunit of RNA polymerase II elongation factor SIII. Proc Natl Acad Sci USA 1994; 91: 5237–5241.
Garrett KP, Aso T, Bradsher JN, Foundling SI, Lane WS, Conaway RC et al. Positive regulation of general transcription factor SIII by a tailed ubiquitin homolog. Proc Natl Acad Sci USA 1995; 92: 7172–7176.
Duan DR, Pause A, Burgess WH, Aso T, Chen DYT, Garrett KP et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science 1995; 269: 1402–1406.
Aso T, Haque D, Barstead RJ, Conaway RC, Conaway JW . The inducible elongin A elongation activation domain: structure, function and interaction with the elongin BC complex. EMBO J 1996; 15: 5557–5566.
Gerber M, Eissenberg JC, Kong S, Tenney K, Conaway JW, Conaway RC et al. In vivo requirement of the RNA polymerase II elongation factor Elongin A for proper gene expression and development. Mol Cell Biol 2004; 24: 9911–9919.
Gerber M, Tenney K, Conaway JW, Conaway RC, Eissenberg JC, Shilatifard A . Regulation of heat shock gene expression by RNA polymerase II elongation factor, Elongin A. J Biol Chem 2005; 280: 4017–4020.
Robertson EJ In: Robertson EJ (ed). Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. IRL Press: Oxford, 1987.
Yamazaki K, Aso T, Ohnishi Y, Ohno M, Tamura K, Shuin T et al. Mammalian Elongin A is not essential for cell viability but is required for proper cell cycle progression with limited alteration of gene expression. J Biol Chem 2003; 278: 13585–13589.
Aso T, Amimoto K, Takebayashi S, Okumura K, Hatakeyama M . Structural organization and chromosome location of the mouse Elongin A gene (Tceb3). Cytogenet Cell Genet 1999; 86: 259–262.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–9367.
Semenza GL, Agani F, Iyer N, Kotch L, Laughner E, Leung S et al. Regulation of cardiovascular development and physiology by hypoxia-inducible factor 1. Ann NY Acad Sci 1999; 874: 262–268.
Harris AL . Hypoxia–A key regulatory factor in tumor growth. Nat Rev Cancer 2001; 2: 38–47.
Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 2002; 10: 995–1005.
Stein S, Thomas EK, Herzog B, Westfall MD, Rocheleau JV, Jackson II RS et al. NDRG1 is necessary for p53-dependent apoptosis. J Biol Chem 2004; 279: 48930–48940.
Hammond EM, Giaccia AJ . The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Commun 2005; 331: 718–725.
Ljungman M, O'Hagan HM, Paulsen MT . Induction of ser15 and lys382 modifications of p53 by blockage of transcription elongation. Oncogene 2001; 20: 5964–5971.
Ben-Porath I, Weinberg RA . When cells get stressed: an integrative view of cellular senescence. J Clin Invest 2004; 113: 8–13.
Han EKH, Ng SC, Arber N, Begemann M, Weinstein IB . Roles of cyclin D1 and related genes in growth inhibition, senescence and apoptosis. Apoptosis 1999; 4: 213–219.
Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol 2002; 22: 3389–3403.
Iwasa H, Han J, Ishikawa F . Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003; 8: 131–144.
Nebreda AR, Porras A . p38 MAP kinases: beyond the stress response. Trends Biochem Sci 2000; 25: 257–260.
Murphy PJM, Galigniana MD, Morishima Y, Harrell JM, Kwok RPS, Ljungman M et al. Pifithrin-α inhibits p53 signaling after interaction of the tumor suppressor protein with hsp90 and its nuclear translocation. J Biol Chem 2004; 279: 30195–30201.
Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R . Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 2003; 34: 263–264.
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev 2000; 14: 34–44.
Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O, Chung DC . Hypoxia-inducible factor-1-independent regulation of vascular endothelial growth factor by hypoxia in colon cancer. Cancer Res 2004; 64: 1765–1772.
Leonard MO, Cottell DC, Godson C, Brady HR, Taylor CT . The role of HIF-1α in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J Biol Chem 2003; 278: 40296–40304.
Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ 2001; 8: 367–376.
Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 2002; 22: 2283–2293.
Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 1993; 8: 3313–3322.
Acknowledgements
We thank T Kamura for plasmids and helpful discussions and A Tsutsui, M Tsuno, K Sugahara, J Piao and K Shiraishi for their technical assistance. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and the Yamanouchi Foundation for Research on Metabolic Disorders.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Oren
Rights and permissions
About this article
Cite this article
Miyata, K., Yasukawa, T., Fukuda, M. et al. Induction of apoptosis and cellular senescence in mice lacking transcription elongation factor, Elongin A. Cell Death Differ 14, 716–726 (2007). https://doi.org/10.1038/sj.cdd.4402067
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402067
Keywords
This article is cited by
-
Downregulation of ARID1A in gastric cancer cells: a putative protective molecular mechanism against the Harakiri-mediated apoptosis pathway
Virchows Archiv (2021)
-
Multiple across-strain and within-strain QTLs suggest highly complex genetic architecture for hypoxia tolerance in channel catfish
Molecular Genetics and Genomics (2017)
-
Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1
The EMBO Journal (2008)
-
Role of EndoG in development and cell injury
Cell Death & Differentiation (2007)
