
Abstract
Full and productive activation of T lymphocytes relies on the simultaneous delivery of T cell receptor (TCR)- and coreceptor-derived signals. In naïve T cells engagement of the TCR alone causes anergy, while TCR triggering of preactivated T cells results in activation-induced cell death. Costimulatory signals are prominently mirrored by the activation of NF-κB, which needs input from the TCR as well as from coreceptors in order to be fully activated and to fulfil its crucial function in the immune response. Coreceptor-generated signals tightly control the duration and amplitude of the NF-κB response. The activation of IκB kinase (IKK) complex at the contact zone between a T cell and an antigen-presenting cell offers the unique opportunity to study the spatial organization of IKK activation. Recent studies indicate that coreceptor pathways influence the threshold activities of many signalling mediators and thus act on multiple layers of the NF-κB pathway.
Similar content being viewed by others


The Concept of T Cell Costimulation
Antigenic stimulation of naïve T cells results in differentiation to effector and memory cells or alternatively in anergy and apoptosis. Full activation of T cells critically depends on the activation of the T cell receptor (TCR) and CD3 together with a second set of signals delivered by costimulatory receptors, as originally proposed by the ‘two signal hypothesis’.1 The first signal is initiated upon binding of the TCR to a specific antigenic peptide presented by the major histocompatibility complex (MHC) on the surface of an antigen-presenting cell (APC). Although the rate of TCR signalling is also influenced by concentration and affinity of the antigen and the duration of T cell/APC interaction,2 efficient T cell activation requires costimulatory signals derived from cell surface receptors. These coreceptor-driven signals are necessary to boost a productive immune response, which leads to cytokine production, increased survival and clonal expansion of the naïve T cell.3, 4 In contrast, engagement of the TCR in the absence of costimulation induces anergy and thereby promotes T cell tolerance. This dual control system provides a fail-safe mechanism and impedes auto-immune reactions by preventing T cell activation by ‘unauthorized APCs’ in the absence of costimulatory molecules5 (see also Green DR, this issue of CDD). Furthermore, weak signals derived from the stimulation of only few TCRs can be substantially augmented by costimulatory receptors.
Coreceptors include CD28, cytotoxic T lymphocyte antigen 4 (CTLA4), inducible T cell costimulator (ICOS), programmed cell death protein 1 (PD-1) and CD7.6, 7 All of these proteins belong to the immunoglobulin supergene family and share some signalling motifs contained in their cytoplasmic domains. The different receptors are expressed on different subsets of T cells and can function as costimulators (CD28, ICOS and CD7) or corepressors (CTLA4, PD-1) of T cell activation. CD28 is expressed by activated and naïve T cells, making this coreceptor especially important for the induction of primary immune responses. In contrast, ICOS and CTLA4 are only expressed on activated and memory T cells and are more relevant for regulation of the T helper type 2 (Th2) responses and the termination of T cell activation, respectively. Different coreceptors can be engaged by overlapping ligands, as in the case for CD28 and CTLA4, which are competing for association of B7-1 (CD80) or B7-2 (CD86) on APCs.8 Other coreceptors are triggered by distinct ligands, for example, B7H for ICOS, PD-1L for PD-1 and K12 for CD7.9 In addition to the CD28 family of costimulatory receptors, TCR-mediated activation and proliferation can be modulated by chemokine receptors10 and some tumor necrosis factor (TNF) family members.11
Given the great amount of genetic and biochemical data obtained for the CD28 receptor, this review will mainly discuss the role of CD28 for costimulation and its effects on NF-κB activity. CD28-mediated T cell costimulation lowers the threshold for TCR-derived signals, enhances the production of growth-stimulatory lymphokines such as interleukin-2 (IL-2), and activates the cytolytic potential of cytotoxic T cells. Furthermore, costimulation is critical for T cell-dependent antibody production and is required for germinal center formation.12 Accordingly, CD28-deficient mice show strongly impaired responses to immune challenges such as a compromised Th2 development, immunoglobulin class switching and an increased susceptibility to various infectious pathogens.13 The paramount importance of CD28 for the efficient functioning of the T cell response is also reflected by the finding that some inflammatory diseases are associated with specific CD28 mutations.14
The scientific literature reflects a long debate on the question whether the TCR and the coreceptors induce separate signal pathways (qualitative model) or whether the signalling routes employed by both receptor systems are entirely overlapping (quantitative model).13 The truth probably lies somewhere in between, as we shall discuss here the occurrence of ‘CD28 only’ signals, but also signalling pathways shared between the TCR and coreceptors. In addition, the prevalence of quantitative or qualitative signalling depends on the biological situation. For example, the qualitative model will be more relevant for the situation of trans signalling, where the TCR and the coreceptor are triggered separately by distinct cells, a major mechanism of tissue rejection.15
Modulation of NF-κB has been observed for most coreceptor pathways and thus the IκB kinase complex (IKK)/NF-κB signalling cascade is thought to critically contribute to integrate TCR and costimulatory signals. The mammalian NF-κB family consists of five different DNA-binding subunits: NF-κB1 (p105 and p50), NF-κB2 (p100 and p52), c-Rel, RelB and RelA (p65). NF-κB family members share an N-terminal Rel homology domain (RHD) that mediates DNA binding, dimerization, nuclear translocation and interaction with the inhibitory IκB proteins, which retain NF-κB dimers in the cytoplasm.16 A multitude of extracellular signals induces NF-κB activation, which plays a prominent role in diverse physiological conditions, such as mounting an immune response, induction of an inflammatory or pathogenic state or activation of morphogenic and developmental processes. NF-κB can be activated by two pathways: The canonical pathway is elicited by many agents, including microbial pathogens, proinflammatory cytokines or T cell costimulation, which ultimately lead to activation of the IκB kinase (IKK) complex, concomitant phosphorylation and degradation of small cytosolic IκB proteins and activation of RelA- and c-Rel-containing heterodimers.17, 18 The noncanonical pathway leads to processing of NF-κB2 and the selective activation of p52/RelB dimers, and is important for survival of premature B cells and development of secondary lymphoid organs.19 Upon their release from inhibitory IκBs, NF-κB dimers translocate to the nucleus, where they bind to their cognate DNA sequence and regulate transcription of diverse genes encoding growth factors, cytokines, cell adhesion molecules, and pro- and antiapoptotic proteins. All current evidence indicate that TCR/CD28 costimulation activates the canonical, but not the noncanonical, NF-κB pathway.20 Even though many of the key molecules that regulate TCR-mediated IKK/NF-κB activation have been identified (see reviews of Israel and Tschopp in this issue of CDD), the underlying molecular mechanisms for the integration of positively or negatively acting coreceptor pathways are just emerging.
An Important Role of NF-κB for CD28-Mediated Effects
T cell costimulation to NF-κB serves as a paradigm for a signalling system that requires input from the TCR as well as the CD28 system to be fully activated.21 Most T cell autonomous effects of CD28 signalling are under tight regulation of NF-κB, as, for example, the prosurvival function of CD28 involves the NF-κB-dependent upregulation of Bcl-xL22 and the inhibition of p73 expression.23 Similarly, NF-κB proteins are important for the CD28-mediated induction of many chemokines and lymphokines such as IL-2. In addition, CD28 is a master switch that allows T cells to exit the G0 phase and to enter the cell cycle using a cdk6/4-cyclin D-dependent step24 that receives regulatory input from the NF-κB system.25 There is also emerging evidence that NF-κB is involved in the CD28-induced regulation of chromatin modification and architecture. The NF-κB DNA-binding subunit c-Rel is selectively required for CD28-induced chromatin remodelling and nucleosome positioning across the IL-2 promoter, while p65 does not participate in this process.26 CD28-induced expression of Bcl-xL and IL-8 is mediated by the early and selective recruitment of NF-κB p65/p52 DNA-binding subunits to their promoter regions.27 In addition, NF-κB is required for CD28-triggered hyperacetylation of histones H3 and H4 at the IL-5 gene locus.28 It will be interesting to learn whether the reported CD28-mediated loss of cytosine methylation and increase in histone acetylation29 also involve the participation of NF-κB family members and what cellular pathways mediate CD28-triggered and NF-κB-dependent chromatin remodelling.
Coreceptor Architecture and Clustering
Activation of T cells is initiated by the formation of a T cell/APC conjugate that involves the formation of an immunological synapse (IS) at the cell/cell interphase. Proteins segregate into two distinct regions within this lipid raft-rich contact zone: a central area, referred to as the central supramolecular activation cluster (c-SMAC), and the peripheral region called the p-SMAC.30 The c-SMAC contains the TCR and also CD28, the p-SMAC is enriched in leukocyte function-associated antigen 1 (LFA1). LFA1 binds to its ligand intercellular adhesion molecule (ICAM) expressed on the APC surface and thus contributes to the stabilization of the APC/T cell complex.31
The costimulatory CD28, ICOS and CTLA-4 receptors are homodimers consisting of extracellular paired variable domain-like immunoglobulin domains linked to single transmembrane domains and a cytoplasmic portion that harbors an array of signalling motifs.32 The recent elucidation of the CD28 extracellular domain structure in complex with the Fab fragment of a mitogenic antibody reveals insight into the mode of CD28 receptor activation.33 The structure of monomeric CD28 most closely resembles that of monomeric CTLA-4, but their mode of ligand binding seems to be different. Whereas the interactions of CD80 and CTLA-4 are each bivalent, interactions involving CD28 and CD86 are monovalent.34 This implies that CD28-mediated signalling employs a mechanism that does not depend on induced receptor aggregation, as it occurs, for example, in the case of TNF receptors. CD28 receptor binding might generate signals upon forcing a change in the relative monomer orientation of dimeric CD28, or affect the size-dependent segregation of signalling molecules in a way analogous to that proposed for TCR triggering by endogenous ligands.35
Triggering of the CD28 receptor allows its recruitment to the c-SMAC by a complex mechanism that also employs the dynamic formation of protein/protein networks.36 This recruitment allows CD28 tyrosine phosphorylation by the c-SMAC kinase Lck,37 thus creating docking sites for interacting proteins. CD28 accumulates in the synapse at very early time points at the onset of the calcium signal. While some reports show a role of CD28 for the formation and stabilization of the mature immunologic synapse and c-SMAC localization of PKCθ,38, 39 quantitative imaging failed to substantiate these findings.40 Life cell imaging approaches showed a surprisingly complex and highly dynamic shuttling of NF-κB activating signalling molecules between the c-SMAC and punctuated structures in the cytosol,41 but the nature and function of these structures need further investigations.
All B7 family coreceptors have relatively short cytoplasmic domains, as displayed for CD28 in Figure 1. The various coreceptors share an YXXM motif, allowing the inducible interaction with the p85 adaptor subunit of class Ia phosphatidylinositol 3-kinase (PI3K). Phosphorylation of the tyrosine contained in the PI3K recruitment motif allows the interaction of p85 via its most N-terminal Src homology (SH)2 domain. The catalytic subunit of PI3K principally phosphorylates inositol phospholipids on carbon atom 3 to generate the phospholipids PI 3,4-biphosphate (PIP2) and PI 3,4,5-triphosphate (PIP3). In the cytoplasmic domain of CD28, the same YXXM motif is used to recruit the SH2 domain containing adaptor protein growth-factor receptor-bound protein 2 (Grb2) and homologous proteins such as Gads (Grb2-related adaptor protein) and GRID (Grb2-related protein with insert domain).42 The cytoplasmic domain of CD28 also allows binding of the Tec family tyrosine kinases, Itk, which employs its SH3 domain to interact with one or two PXXP sequences. The more C-terminal PYAP sequence is bound by the tyrosine kinase Lck, which phosphorylates the cytoplasmic tail and thus prevents binding of the negatively regulating serine/threonine phosphatase PPA2.43

The cytoplasmic domain of CD28. The entire amino-acid sequence is shown, binding sites for the indicated signalling proteins are marked by underlining
Coreceptor Signalling to IKK/NF-κB
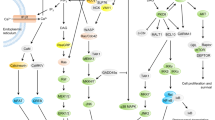
The first connection between NF-κB and CD28 was made by the identification of NF-κB binding to the CD28 response element (CD28RE) in the IL-2 promoter. 44 Stimulation of this coreceptor alone is sufficient to induce weak IKK activation, which allows subsequent phosphorylation of IκB45 and p65.46 However, full NF-κB activation definitively requires input from CD28 as well as from the TCR.45, 47 Here, we will focus on the organization of the early signalling events, as the late steps of signal delivery into the IKK complex are already covered in this issue by the reviews of Israel and Tschopp. Although initiation of the PI3K and Grb2 pathways requires identical structural motifs on the CD28 receptor, we will discuss them separately for the sake of clarity (see Figure 2a and b).

Schematic summary of CD28 signalling. For the sake of simplicity, PI3K-dependent (a) and -independent (b) signalling pathways are displayed separately. PIP2 and PIP3 are shown in green, further details are given in the text
PI3K-dependent NF-κB activation
Recruitment of the p85 regulatory subunit of PI3K to the CD28 receptor is a key event for coupling coreceptors to the IKK/NF-κB signalling pathway (Figure 2a). The p85 subunit mediates two basic effects: Firstly, it allows SH3 domain-dependent binding of Rho, Rac and Cdc42, thus allowing them to contact protein complexes displaying guanine nucleotide exchange factor (GEF) activity and to be loaded with GTP.48 Secondly, p85 docking allows for activation of PI3K kinase activity, which creates PIP2 and PIP3, which are localizing to the inner leaflet of the plasma membrane. The role of PI3K in the delivery of CD28-mediated signals into the NF-κB activation pathway was revealed upon mutation of the PI3K interaction site in the cytosolic tail of CD28. These mutants failed to recruit PKCθ to the c-SMAC and thus to promote NF-κB activation.49 In contrast, mutation of the PI3K interaction site still allowed CD28-mediated stabilization of IL-2 mRNA, showing that both mechanisms are distinctively regulated.
PI3K phosphorylated lipids serve as second messengers that bind various proteins harboring pleckstrin homology (PH) domains, including phosphatidylinositol-dependent kinase 1 (PDK1), protein kinase B (PKB) and the GEF Vav.50 Activated PDK1 signals to several proteins, one of them being the interaction partner and phosphorylation substrate PKCθ,51 an essential component of the antigen receptor-mediated NF-κB activation pathway (see reviews by Israel and Tschopp). Silencing of PDK1 expression by siRNA prevents receptor-induced IKK lipid raft recruitment and NF-κB activation. PDK1-mediated IKK activation also proceeds by a parallel pathway that employs an interaction between PDK1 and Carma1. This interaction feeds signals into the Carma1/Bcl10/MALT1 complex and thus might potentially enhance TRAF6-mediated NEMO/IKKγ ubiquitination and subsequent IKK activation51 (Chen, this issue of CDD). However, the generation and analysis of PDK1-deficient primary T lymphocytes will be needed to clarify the role of this kinase for IKK/NF-κB activation in vivo.
PDK1 can also catalyze the phosphorylation of PKB/Akt, which is activated by CD28-derived signals.52 The occurrence of this kinase in three isoforms has thus far precluded the analysis of its relevance for NF-κB signalling by genetic inactivation. However, transgenic mice expressing a constitutively active form of PKB show an increased NF-κB activation in response to CD28 ligation,53 suggesting its importance for NF-κB activation. Among the prominent phosphorylation targets of PKB/Akt is glycogen synthase kinase 3 (GSK3)β, which is phosphorylated and thus inactivated in response to CD28 triggering.54 While the role of GSK3 in the regulation of glycogen synthase, protein translation and the nuclear localization of nuclear factor of activated T cells (NFAT) is well established, its importance for NF-κB activation in response to T cell costimulation remains elusive and awaits the availability of conditional or cell specific knockout mice. PKB/Akt-derived signals to NF-κB can also travel on a pathway that employs its interaction with the serine/threonine kinase Cot/Tpl-2. PKB/Akt-mediated phosphorylation of Cot/Tpl-2 at serine 400 is required for its function to stimulate IKK activity.55 Cot/TPL-2-mediated IKK activation depends on NF-κB-inducing kinase (NIK),56 which is functional for costimulation-induced NF-κB activation, as revealed by the analysis of aly mice containing a mutant NIK protein.57 Costimulation-induced NF-κB activation also involves the kinases MEKK158 and the Bcl10 interacting kinase RIP2.59 However, the exact molecular targets for most of these kinases feeding into the CD28-triggered IKK/NF-κB pathway remain unresolved.
PI3K-independent NF-κB activation via Grb2 and Vav
Although the relevance of PI3K for coreceptor function is beyond controversy, other PI3K-associated receptors cannot substitute for CD28 signalling, which shows the involvement of further pathways. An example for this situation is provided by the ICOS coreceptor, which employs the YMFM motif to recruit PI3K. However, this sequence fails to bind to the adaptor protein Grb2 and to mediate NF-κB activation and IL-2 transcription. Point mutation of this ICOS coreceptor docking site to the YMNM sequence found in the CD28 tail allows Grb2 recruitment and activation of transcription from NF-κB and the CD28RE containing promoter,60 providing an elegant proof for the involvement of this adaptor protein. Grb2 lacks any obvious enzymatic activity, but contains one SH2 and two SH3 domains, which allow the versatile wiring to other signalling mediators.
The interaction of Grb2 and the Vav protein is a crucial step for T cell costimulation (Figure 2b). The Vav protein family comprises three members: Vav1, Vav2 and Vav3.61 Most knowledge has been gathered for the Vav1 protein (initially termed Vav), which is exclusively expressed in the hematopoietic lineage. Vav1 contains a complex arrangement of domains, including a PH domain that most probably binds to PIP2 and PIP3. Vav also contains a Dbl homology (DH) region, which allows this protein to function as a GEF for Rho-family GTPases.62 The multi-domain Vav protein is kept inactive in unstimulated cells by intramolecular interactions. Phosphorylation of Vav1 allows conformational changes and the induction of GEF.61
Vav1-deficient T cells show strongly impaired IκBα degradation and NF-κB DNA binding in response to T cell costimulation.63 The observed residual NF-κB activation is most probably due to the compensatory function of Vav2 and Vav3, as documented in Vav1/2/3-null mice.64 Reconstitution experiments in Vav1-deficient Jurkat T cells showed that only the wild-type form, but not a Vav1 point mutant lacking the GEF activity, was able to rescue the NF-κB response.65 Costimulation results in a prolonged and sustained tyrosine phosphorylation and membrane localization of Vav1 in comparison to TCR activation alone.66 These findings suggest that Vav1 functions as a signal integrator that receives input from the CD28 signalling system as well as from the TCR. The activated Vav1 protein distributes signals into many directions: The GEF activity triggers the function of Rac and Rho, thus reorganizing the actin cytoskeleton.67 In addition, Vav1 binds constitutively to PKCθ68 and overexpression experiments show a powerful synergism of both proteins for NF-κB activation.69 However, the molecular mechanisms relaying Vav1-derived signals to PKCθ remain to be elaborated, as the effect of Vav1 deficiency on recruitment of PKCθ into the cSMAs remains controversial.65, 67
In addition, costimulation facilitates the interaction of Vav1 with further proteins such as SLP-76. CD28 receptor stimulation induces the formation of a membrane-associated Vav1/SLP-76 complex. Cooperation between SLP-76 and Vav1 for the induction of IL-2 production depends on the mutual interaction of both proteins, as SLP-76 point mutants unable to contact Vav1 fail to sustain Vav1 activity.70 The analysis of SLP-76-deficient Jurkat cells revealed the importance of SLP-76 for TCR/CD28-stimulated IκBα degradation and NF-κB activation,71 as well a defective lipid raft recruitment of PKCθ and the IKK complex.69 SLP-76 is also required for optimal tyrosine phosphorylation and activation of phospholipase C-γ1 (PLCγ),72 which converts PIP2 to inositol (1,4,5)-triphosphate (IP3) and the PKC activator diacylglycerol. In mature T cells, activated PKCθ submits the signals towards the IKK complex.73
Vav1 is also a target for signals emanating from the CD28 receptor alone in the absence of TCR engagement. CD28 stimulation induces a rapid but transient dissociation of Vav1 and PKCθ,69 and causes an arginine methylation of nuclear Vav1.74 Even though these results prove that CD28 can have a qualitative impact on T cell signalling, the molecular mechanisms, functional consequences and possible connection of these ‘CD28 only’ signals remain to be identified.
The cytoplasmic tail of CD28 still hides some secrets. As discussed above, the YMNM motif nucleates both the activation of the PI3K-dependent as well as the Grb2/Vav-mediated signalling pathway. Domain swapping experiments show that the introduction of the YMNM motif in the cytoplasmic domain of ICOS allows for NF-κB activation, but full and potent NF-κB induction relies on CD28 sequences outside the YMNM motif.60 The CD28 cytoplasmic domain may also bind to further proteins.75, 76 Their identification and the elucidation of the involved mechanisms may be facilitated by the recent availability of superagonistic CD28 antibodies, which bypass the requirement of TCR signals for NF-κB activation.77
CD28 Sets the Threshold: The Role of Ubiquitin Ligases
T cell activation is tightly balanced by positive and negative regulatory mechanisms. Recent studies have highlighted a crucial role of E3 ubiquitin ligases for these processes. E3 ligases catalyze the conjugation of ubiquitin side chains to lysine residues on substrate proteins, which, depending on the type and complexity of the chain linkage, can target cellular proteins for proteasomal degradation or alter their function or subcellular distribution. The large and structurally heterogeneous group of regulatory E3s is composed of more than 500 members that can be classified into two major classes: the really interesting new gene (RING) and homology to E6-AP C terminus (HECT) E3 ligases.78
Triggering of the TCR in the absence of costimulatory signals induces anergy, which is even the default pathway for antigen-experienced CD4+ T cell clones. Anergic cells fail to activate NF-κB and synthesize IL-2 even after appropriate restimulation and thus fail to proliferate and differentiate (see review in this issue by DR Greene). The process of anergy induction can be subdivided into two periods (Figure 3a and b). In an initiation period, TCR signalling without costimulation evokes a PLCγ-dependent and sustained increase in cytosolic Ca2+ concentrations and the induction of NFAT-dependent transcription.79 The NFAT target genes include the RING E3 ligases Cbl-b and GRAIL, as well as the HECT E3 Itch and Nedd4.80 TCR/CD28 restimulation of ‘TCR-only’ treated T cells promotes the execution of the anergic gene program. Just like in naïve T cells upon a first encounter with an antigen, primary SMAC formation is not affected in anergic T cells, but the SMACs are highly unstable and the synapse collapses within minutes after engagement.80 PLCγ and PKCθ, two key T cell signalling molecules, are degraded in anergic T cells, and the relative contribution of this mechanism is evident from the analysis of T cells derived from Cbl-b and Itch-deficient mice, which show a stabilization of PLCγ and PKCθ and accordingly are more resistant to anergy induction. Also, the Bcl10 protein is proteolytically eliminated by a similar mechanism, ensuring the selective termination of NF-κB signalling in response to T cell costimulation.81 Notably, removal of PLCγ, PKCθ and Bcl10 proteins involves trafficking to the lysosomal compartment rather than proteasomal degradation. While it is clear that during the initiation period calcineurin/NFAT activity is sufficient for the induction of E3 ligase expression, it remains to be determined what prevents the induction of the anergic gene program in response to costimulation (Figure 3c). One candidate is NF-κB, and it can be expected that future studies unravel more fascinating mechanisms mediating the mutual interplay between NF-κB and coreceptor signalling.

A model for threshold regulation by anergy-induced ubiquitination. (a) Stimulation of the TCR in the absence of costimulation is sufficient to induce expression of the ubiquitin E3 ligases Itch, Cbl-b and Grail. (b) Costimulation of anergic T cells containing these upregulated E3s leads to their activation and the subsequent lysosomal elimination of PLCγ and PKCθ. (c) Costimulation of T cells allows for full activation of signalling pathways and inhibits expression of the anergy E3 ligases by an unknown mechanism. Active signalling elements are shown in black, inactive pathways are shown in gray
The role of these E3 ligases in the regulation of molecular thresholds is also evident from mouse models. Cbl-b-deficient T cells do not require CD28 engagement for IL-2 production and proliferation, and Cbl-b seems to regulate the CD28-dependent activation of Vav1 in T cells.82 The proline-rich sequence of Cbl-b interacts with the SH3 domain of the p85 regulatory subunit of PI3K and enhances ubiquitination of p85.83 Without affecting the stability of p85, Cbl-b impedes association of p85 to CD28, but also to TCRζ by a yet unknown mechanism. Cbl-b deficiency can rescue some of the defects of CD28−/− mice, for example, Vav1 phosphorylation and proliferation.84 Cbl-b also cooperates with c-Cbl in downmodulating the TCR after its engagement and thus directly affects TCR signalling.85 The actual effects of Cbl-b on downstream effector pathways such as IKK or c-Jun N-terminal kinase (JNK) signalling still need to be investigated. In another study, Cbl-b-induced ubiquitination of PLCγ interferes with TCR signalling in anergic cells by diminishing phosphorylation of PLCγ without affecting its stability.86 In addition, Cbl-b amounts are not only determined by transcriptional upregulation, but also by coreceptor-mediated stabilization events. While CD28 signalling results in increased Cbl-b ubiquitination/degradation, activation of CTLA-4 signalling increases the amount of Cbl-b.87
The E3 ligase Itch can not only catalyze ubiquitination and degradation of PLCγ and Bcl10, but also causes depletion of the AP-1 family members JunB and c-Jun upon TCR/CD28 engagement.88 Thus, Itch can interfere at multiple levels with lymphocyte activation, which is in agreement with the profound immunological disorders that are caused through ablation of Itch in mice.89, 90 In this respect, it is interesting to note that Itch activity can be modulated by JNK, which indicates a novel function of JNK as a negative regulator of T cell signalling.88
Not only do CD28-derived signals impinge on the expression and activity of ubiquitin E3 ligases, but also ICOS-mediated signalling is repressed by the RING-type ubiquitin ligase Roquin.91 Sanroque (san/san) mice, which carry a homozygous mutation in the roquin gene, develop an autoimmune phenotype that resembles human systemic erythematosus and display a substantial increase in previously activated memory T cells. On a molecular basis, ICOS expression is greatly enhanced after TCR engagement in san/san mice, which suggests that this coreceptor is a target for the ubiquitin ligase Roquin. Besides its RING domain, Roquin contains an RNA-binding CCCH zinc-finger. ICOS induction is observed at the mRNA and protein levels, which led the authors to suggest a model by which Roquin regulates ICOS expression by affecting mRNA stability and/or translation. Certainly additional data are required to validate this type of regulation, but it is conceivable that the mutation in Roquin and the increase in ICOS expression lower the threshold for other costimulatory molecules like CD28 and thereby increase the numbers of self-reactive T cells.
Outlook and Therapeutic Implications
Great progress has been made in the identification of signalling molecules responsible for the delivery of signals from T cell coreceptors, but many fundamental questions remain to be answered. Structure–function analysis shows that the different domains in the CD28 cytoplasmic tail act redundantly to induce IL-2 production and proliferation of CD4+ T cells. However, CD28-mediated IL-4 production and Th2 differentiation are dependent on the cooperative activity of at least two structural motifs within the cytoplasmic tail, indicating that distinct mechanisms and signalling pathways contribute to the various responses by CD28.92 Site-directed mutagenesis of amino acids in the cytoplasmic tails and domain-swapping experiments will remain valuable tools to define the molecular events critical for coreceptor function and downstream responses such as NF-κB activation. Membrane microdomains have been implicated in proximal T cell signalling, but their origin and properties remain an open issue. Two colour-imaging analyses suggest that the microdomains are created by protein–protein interactions rather than by lipid rafts or an underlying actin cytoskeleton.36 Two-photon microscopy and tracking of single molecules will be needed to pursue the origin and the function of these protein interaction networks in T cell costimulation under physiological conditions. The molecular events allowing coreceptor-mediated chromatin reorganization and protein methylation are not clear and open a new avenue for future research. Novel approaches point to a major contribution of ubiquitin and/or ubiquitin-like modifications for T cell activation and the induction of anergy. New E3 ligases and their respective cellular targets need to be identified to clarify the role of ubiquitin linkage in T cell signalling (e.g. degradation versus signal propagation) and to substantiate their potential functions in the regulation of positive and negative signalling circuits in T cells. Owing to their importance in mounting a productive immune response, T cell costimulatory receptors are attractive targets for therapeutic intervention. The blockage of costimulatory functions by CTLA-4-Ig is in phase 3 clinical trials for the treatment of rheumatoid arthritis.93 In addition, augmentation of costimulation by superagonistic antibodies to enhance the immune response and to combat cancers that escaped from the immune system is currently under investigation in clinical trials.94 Future studies must determine whether the activation of negative regulatory feedback mechanisms, such as the depletion of signalling mediators by ubiquitin ligases, will be a valuable tool for the treatment of autoimmune diseases or to counteract tissue rejection.
Abbreviations
- APC:
-
antigen-presenting cell
- CTLA4:
-
cytotoxic T lymphocyte antigen 4
- GEF:
-
guanine exchange factor
- GSK3:
-
glycogen synthase kinase 3
- HECT:
-
homology to E6-AP C terminus
- ICAM:
-
intercellular adhesion molecule
- ICOS:
-
inducible T cell costimulator
- IKK:
-
IκB kinase
- IL-2:
-
interleukin-2
- IS:
-
immunological synapse
- JNK:
-
c-Jun N-terminal kinase
- LFA1:
-
leukocyte function-associated antigen 1
- MHC:
-
major histocompatibility complex
- NIK:
-
NF-κB-inducing kinase
- PD-1:
-
programmed cell death protein 1
- PDK1:
-
phosphatidylinositol-dependent kinase 1
- PH:
-
pleckstrin homology
- PI3K:
-
phosphatidylinositol 3-kinase
- PIP2:
-
phosphatidylinositol 3,4-biphosphate
- PIP3:
-
phosphatidylinositol 3,4,5-triphosphate
- PKB:
-
protein kinase B
- PLCγ:
-
phospholipase C-γ1
- RHD:
-
Rel homology domain
- RING:
-
really interesting new gene
- SH:
-
Src homology
- SMAC:
-
supramolecular activation cluster
- TCR:
-
T cell receptor
- Th2:
-
T helper type 2
- TNF:
-
tumor necrosis factor
References
Bretscher PA (1999) A two-step, two-signal model for the primary activation of precursor helper T cells. Proc. Natl. Acad. Sci. USA 96: 185–190
Gett AV, Sallusto F, Lanzavecchia A and Geginat J (2003) T cell fitness determined by signal strength. Nat. Immunol. 4: 355–360
Appleman LJ and Boussiotis VA (2003) T cell anergy and costimulation. Immunol. Rev. 192: 161–180
Frauwirth KA and Thompson CB (2002) Activation and inhibition of lymphocytes by costimulation. J. Clin. Invest. 109: 295–299
Schwartz RH (1990) A cell culture model for T lymphocyte clonal anergy. Science 248: 1349–1356
Rudd CE and Schneider H (2003) Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat. Rev. Immunol. 3: 544–556
Sharpe AH and Freeman GJ (2002) The B7-CD28 superfamily. Nat. Rev. Immunol. 2: 116–126
Pentcheva-Hoang T, Egen JG, Wojnoonski K and Allison JP (2004) B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21: 401–413
Greenwald RJ, Freeman GJ and Sharpe AH (2005) The B7 family revisited. Annu. Rev. Immunol. 23: 515–548
Molon B, Gri G, Bettella M, Gomez-Mouton C, Lanzavecchia A, Martinez AC, Manes S and Viola A (2005) T cell costimulation by chemokine receptors. Nat. Immunol. 6: 465–471
Watts TH (2005) TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 23: 23–68
Ferguson SE, Han S, Kelsoe G and Thompson CB (1996) CD28 is required for germinal center formation. J. Immunol. 156: 4576–4581
Acuto O and Michel F (2003) CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat. Rev. Immunol. 3: 939–951
Popat S, Hearle N, Hogberg L, Braegger CP, O'Donoghue D, Falth-Magnusson K, Holmes GK, Howdle PD, Jenkins H, Johnston S, Kennedy NP, Kumar PJ, Logan RF, Marsh MN, Mulder CJ, Torinsson Naluai A, Sjoberg K, Stenhammar L, Walters JR, Jewell DP and Houlston RS (2002) Variation in the CTLA4/CD28 gene region confers an increased risk of coeliac disease. Ann. Hum. Genet. 66: 125–137
Mandelbrot DA, Kishimoto K, Auchincloss Jr. H, Sharpe AH and Sayegh MH (2001) Rejection of mouse cardiac allografts by costimulation in trans. J. Immunol. 167: 1174–1178
Berkowitz B, Huang DB, Chen-Park FE, Sigler PB and Ghosh G (2002) The x-ray crystal structure of the NF-kappa B p50.p65 heterodimer bound to the interferon beta–kappa B site. J. Biol. Chem. 277: 24694–24700
Hayden MS and Ghosh S (2004) Signaling to NF-kappaB. Genes Dev. 18: 2195–2224
Viatour P, Merville MP, Bours V and Chariot A (2005) Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 30: 43–52
Bonizzi G and Karin M (2004) The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25: 280–288
Li Y, Sedwick CE, Hu J and Altman A (2005) Role for protein kinase Ctheta (PKCtheta) in TCR/CD28-mediated signaling through the canonical but not the non-canonical pathway for NF-kappaB activation. J. Biol. Chem. 280: 1217–1223
Kane LP, Lin J and Weiss A (2002) It's all Rel-ative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 23: 413–420
Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T and Thompson CB (1995) CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3: 87–98
Wan YY and DeGregori J (2003) The survival of antigen-stimulated T cells requires NFkappaB-mediated inhibition of p73 expression. Immunity 18: 331–342
Lea NC, Orr SJ, Stoeber K, Williams GH, Lam EW, Ibrahim MA, Mufti GJ and Thomas NS (2003) Commitment point during G0⩾G1 that controls entry into the cell cycle. Mol. Cell. Biol. 23: 2351–2361
Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B and Pestell RG (2001) NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 12: 73–90
Rao S, Gerondakis S, Woltring D and Shannon MF (2003) c-Rel is required for chromatin remodeling across the IL-2 gene promoter. J. Immunol. 170: 3724–3731
Marinari B, Costanzo A, Marzano V, Piccolella E and Tuosto L (2004) CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-kappaB subunits on IL-8 and Bcl-xL gene promoters. Proc. Natl. Acad. Sci. USA 101: 6098–6103
Inami M, Yamashita M, Tenda Y, Hasegawa A, Kimura M, Hashimoto K, Seki N, Taniguchi M and Nakayama T (2004) CD28 costimulation controls histone hyperacetylation of the interleukin 5 gene locus in developing th2 cells. J. Biol. Chem. 279: 23123–23133
Thomas RM, Gao L and Wells AD (2005) Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J. Immunol. 174: 4639–4646
Lin J, Miller MJ and Shaw AS (2005) The c-SMAC: sorting it all out (or in). J. Cell. Biol. 170: 177–182
Kolanus W, Nagel W, Schiller B, Zeitlmann L, Godar S, Stockinger H and Seed B (1996) Alpha L beta 2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell 86: 233–242
Carreno BM and Collins M (2002) The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 20: 29–53
Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, Hunig T, Sorensen P, Stuart DI and Davis SJ (2005) Crystal structure of a soluble CD28–Fab complex. Nat. Immunol. 6: 271–279
Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA and Davis SJ (2002) The interaction properties of costimulatory molecules revisited. Immunity 17: 201–210
Davis SJ and van der Merwe PA (2003) TCR triggering: co-receptor-dependent or -independent? Trends Immunol. 24: 624–626; author reply 626–627
Douglass AD and Vale RD (2005) Single-molecule microscopy reveals plasma membrane microdomains created by protein–protein networks that exclude or trap signaling molecules in T cells. Cell 121: 937–950
Sadra A, Cinek T and Imboden JB (2004) Translocation of CD28 to lipid rafts and costimulation of IL-2. Proc. Natl. Acad. Sci. USA 101: 11422–11427
Andres PG, Howland KC, Dresnek D, Edmondson S, Abbas AK and Krummel MF (2004) CD28 signals in the immature immunological synapse. J. Immunol. 172: 5880–5886
Huang J, Lo PF, Zal T, Gascoigne NR, Smith BA, Levin SD and Grey HM (2002) CD28 plays a critical role in the segregation of PKC theta within the immunologic synapse. Proc. Natl. Acad. Sci. USA 99: 9369–9373
Burack WR, Lee KH, Holdorf AD, Dustin ML and Shaw AS (2002) Cutting edge: quantitative imaging of raft accumulation in the immunological synapse. J. Immunol. 169: 2837–2841
Schaefer BC, Kappler JW, Kupfer A and Marrack P (2004) Complex and dynamic redistribution of NF-kappaB signaling intermediates in response to T cell receptor stimulation. Proc. Natl. Acad. Sci. USA 101: 1004–1009
Ellis JH, Ashman C, Burden MN, Kilpatrick KE, Morse MA and Hamblin PA (2000) GRID: a novel Grb-2-related adapter protein that interacts with the activated T cell costimulatory receptor CD28. J. Immunol. 164: 5805–5814
Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ and Thompson CB (2000) The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 13: 313–322
Verweij CL, Geerts M and Aarden LA (1991) Activation of interleukin-2 gene transcription via the T-cell surface molecule CD28 is mediated through an NF-kB-like response element. J. Biol. Chem. 266: 14179–14182
Harhaj EW and Sun SC (1998) IkappaB kinases serve as a target of CD28 signaling. J. Biol. Chem. 273: 25185–25190
Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M and Schmitz ML (2004) Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 172: 6336–6344
Weil R, Schwamborn K, Alcover A, Bessia C, Di Bartolo V and Israel A (2003) Induction of the NF-kappaB cascade by recruitment of the scaffold molecule NEMO to the T cell receptor. Immunity 18: 13–26
Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP and Scita G (2003) Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J. Cell. Biol. 160: 17–23
Sanchez-Lockhart M, Marin E, Graf B, Abe R, Harada Y, Sedwick CE and Miller J (2004) Cutting edge: CD28-mediated transcriptional and posttranscriptional regulation of IL-2 expression are controlled through different signaling pathways. J. Immunol. 173: 7120–7124
Lemmon MA (2005) Pleckstrin homology domains: two halves make a hole? Cell 120: 574–576
Lee KY, D'Acquisto F, Hayden MS, Shim JH and Ghosh S (2005) PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science 308: 114–118
Parry RV, Reif K, Smith G, Sansom DM, Hemmings BA and Ward SG (1997) Ligation of the T cell co-stimulatory receptor CD28 activates the serine–threonine protein kinase protein kinase B. Eur. J. Immunol. 27: 2495–2501
Jones RG, Parsons M, Bonnard M, Chan VS, Yeh WC, Woodgett JR and Ohashi PS (2000) Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivo. J. Exp. Med. 191: 1721–1734
Diehn M, Alizadeh AA, Rando OJ, Liu CL, Stankunas K, Botstein D, Crabtree GR and Brown PO (2002) Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl. Acad. Sci. USA 99: 11796–11801
Kane LP, Mollenauer MN, Xu Z, Turck CW and Weiss A (2002) Akt-dependent phosphorylation specifically regulates Cot induction of NF-kappa B-dependent transcription. Mol. Cell. Biol. 22: 5962–5974
Lin X, Cunningham Jr. ET, Mu Y, Geleziunas R and Greene WC (1999) The proto-oncogene Cot kinase participates in CD3/CD28 induction of NF-kappaB acting through the NF-kappaB-inducing kinase and IkappaB kinases. Immunity 10: 271–280
Matsumoto M, Yamada T, Yoshinaga SK, Boone T, Horan T, Fujita S, Li Y and Mitani T (2002) Essential role of NF-kappa B-inducing kinase in T cell activation through the TCR/CD3 pathway. J. Immunol. 169: 1151–1158
Tao L, Wadsworth S, Mercer J, Mueller C, Lynn K, Siekierka J and August A (2002) Opposing roles of serine/threonine kinases MEKK1 and LOK in regulating the CD28 responsive element in T-cells. Biochem. J. 363: 175–182
Ruefli-Brasse AA, Lee WP, Hurst S and Dixit VM (2004) Rip2 participates in Bcl10 signaling and T-cell receptor-mediated NF-kappaB activation. J. Biol. Chem. 279: 1570–1574
Harada Y, Ohgai D, Watanabe R, Okano K, Koiwai O, Tanabe K, Toma H, Altman A and Abe R (2003) A single amino acid alteration in cytoplasmic domain determines IL-2 promoter activation by ligation of CD28 but not inducible costimulator (ICOS). J. Exp. Med. 197: 257–262
Tybulewicz VL (2005) Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 17: 267–274
Adams JM, Houston H, Allen J, Lints T and Harvey R (1992) The hematopoietically expressed vav proto-oncogene shares homology with the dbl GDP–GTP exchange factor, the bcr gene and a yeast gene (CDC24) involved in cytoskeletal organization. Oncogene 7: 611–618
Costello PS, Walters AE, Mee PJ, Turner M, Reynolds LF, Prisco A, Sarner N, Zamoyska R and Tybulewicz VL (1999) The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK, and NF-kappaB pathways. Proc. Natl. Acad. Sci. USA 96: 3035–3040
Fujikawa K, Miletic AV, Alt FW, Faccio R, Brown T, Hoog J, Fredericks J, Nishi S, Mildiner S, Moores SL, Brugge J, Rosen FS and Swat W (2003) Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 198: 1595–1608
Cao Y, Janssen EM, Duncan AW, Altman A, Billadeau DD and Abraham RT (2002) Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. EMBO J. 21: 4809–4819
Hehner SP, Hofmann TG, Dienz O, Droge W and Schmitz ML (2000) Tyrosine-phosphorylated Vav1 as a point of integration for T-cell receptor- and CD28-mediated activation of JNK, p38, and interleukin-2 transcription. J. Biol. Chem. 275: 18160–18171
Villalba M, Bi K, Rodriguez F, Tanaka Y, Schoenberger S and Altman A (2001) Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J. Cell. Biol. 155: 331–338
Kong YY, Fischer KD, Bachmann MF, Mariathasan S, Kozieradzki I, Nghiem MP, Bouchard D, Bernstein A, Ohashi PS and Penninger JM (1998) Vav regulates peptide-specific apoptosis in thymocytes. J. Exp. Med. 188: 2099–2111
Dienz O, Hehner SP, Droge W and Schmitz ML (2000) Synergistic activation of NF-kappa B by functional cooperation between vav and PKCtheta in T lymphocytes. J. Biol. Chem. 275: 24547–24551
Raab M, Pfister S and Rudd CE (2001) CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity 15: 921–933
Herndon TM, Shan XC, Tsokos GC and Wange RL (2001) ZAP-70 and SLP-76 regulate protein kinase C-theta and NF-kappa B activation in response to engagement of CD3 and CD28. J. Immunol. 166: 5654–5664
Yablonski D, Kuhne MR, Kadlecek T and Weiss A (1998) Uncoupling of nonreceptor tyrosine kinases from PLC-gamma1 in an SLP-76-deficient T cell. Science 281: 413–416
Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL and Littman DR (2000) PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404: 402–407
Blanchet F, Cardona A, Letimier FA, Hershfield MS and Acuto O (2005) CD28 costimulatory signal induces protein arginine methylation in T cells. J. Exp. Med. 202: 371–377
Heller M, Watts JD and Aebersold R (2001) CD28 stimulation regulates its association with N-ethylmaleimide-sensitive fusion protein and other proteins involved in vesicle sorting. Proteomics 1: 70–78
Matsumoto A, Dobashi H, Ohnishi H, Tanaka T, Kubota Y, Kitanaka A, Ishida H, Tokuda M, Waki M, Kubo A and Ishida T (2003) Tyrosine phosphorylation of a novel 100-kDa protein coupled to CD28 in resting human T cells is enhanced by a signal through TCR/CD3 complex. Microbiol. Immunol. 47: 63–69
Kerstan A and Hunig T (2004) Cutting edge: distinct TCR- and CD28-derived signals regulate CD95L, Bcl-xL, and the survival of primary T cells. J. Immunol. 172: 1341–1345
Pickart CM (2004) Back to the future with ubiquitin. Cell 116: 181–190
Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC and Rao A (2002) Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109: 719–731
Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML and Rao A (2004) Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 5: 255–265
Scharschmidt E, Wegener E, Heissmeyer V, Rao A and Krappmann D (2004) Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling. Mol. Cell. Biol. 24: 3860–3873
Krawczyk C, Bachmaier K, Sasaki T, Jones RG, Snapper SB, Bouchard D, Kozieradzki I, Ohashi PS, Alt FW and Penninger JM (2000) Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity 13: 463–473
Fang D and Liu YC (2001) Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2: 870–875
Krawczyk CM, Jones RG, Atfield A, Bachmaier K, Arya S, Odermatt B, Ohashi PS and Penninger JM (2005) Differential control of CD28-regulated in vivo immunity by the E3 ligase Cbl-b. J. Immunol. 174: 1472–1478
Naramura M, Jang IK, Kole H, Huang F, Haines D and Gu H (2002) c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3: 1192–1199
Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, Ohashi P, Wada T, Bloom D, Fathman CG, Liu YC and Penninger JM (2004) Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity 21: 167–177
Li D, Gal I, Vermes C, Alegre ML, Chong AS, Chen L, Shao Q, Adarichev V, Xu X, Koreny T, Mikecz K, Finnegan A, Glant TT and Zhang J (2004) Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 173: 7135–7139
Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC and Karin M (2004) Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306: 271–275
Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, Hunter T, Copeland N, Jenkins N and Liu YC (2002) Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat. Immunol. 3: 281–287
Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA and Copeland NG (1998) The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat. Genet. 18: 143–146
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ and Goodnow CC (2005) A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435: 452–458
Andres PG, Howland KC, Nirula A, Kane LP, Barron L, Dresnek D, Sadra A, Imboden J, Weiss A and Abbas AK (2004) Distinct regions in the CD28 cytoplasmic domain are required for T helper type 2 differentiation. Nat. Immunol. 5: 435–442
Ruderman EM and Pope RM (2005) The evolving clinical profile of abatacept (CTLA4-Ig): a novel co-stimulatory modulator for the treatment of rheumatoid arthritis. Arthritis Res. Ther. 7 (Suppl 2): S21–S25
Riley JL and June CH (2005) The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood 105: 13–21
Acknowledgements
This work was supported by the Swiss National Foundation, EU FP6, the International Association for Cancer Research (MLS) and the Deutsche Forschungsgemeinschaft (DK).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Kroemer
Rights and permissions
About this article
Cite this article
Schmitz, M., Krappmann, D. Controlling NF-κB activation in T cells by costimulatory receptors. Cell Death Differ 13, 834–842 (2006). https://doi.org/10.1038/sj.cdd.4401845
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401845
Keywords
This article is cited by
-
A single-amino acid substitution in the adaptor LAT accelerates TCR proofreading kinetics and alters T-cell selection, maintenance and function
Nature Immunology (2023)
-
Contribution of T- and B-cell intrinsic toll-like receptors to the adaptive immune response in viral infectious diseases
Cellular and Molecular Life Sciences (2022)
-
Can Soluble Immune Checkpoint Molecules on Exosomes Mediate Inflammation?
Journal of Neuroimmune Pharmacology (2022)
-
Low Density Granulocytes and Dysregulated Neutrophils Driving Autoinflammatory Manifestations in NEMO Deficiency
Journal of Clinical Immunology (2022)
-
GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis
Genes & Immunity (2016)
