
Abstract
Renewed attention has been given lately to gangliosides and to their function as intracellular messengers of the adaptive responses to stress. Gangliosides are vital components of cell membranes; therefore, deleterious consequences can result from changes in their chemical composition and concentration, that is, membrane dynamics and structure can be altered as can the behavior of other membrane proteins. The importance of gangliosides in human health is evident in neurodegenerative diseases associated with defects in their degradation. As key modulators of intracellular calcium flux, gangliosides are involved in cellular processes downstream of calcium signaling. In this review, we focus on the effect of ganglioside accumulation on the endoplasmic reticulum calcium homeostasis and on the integrity of the mitochondrial membranes. We discuss how these events elicit an apoptotic program that ultimately leads to cell death. Owing to interorganelle crosstalk, these events are not necessarily self-contained, and gangliosides may serve as the common factor.
Similar content being viewed by others

Introduction
Since their discovery in the late 1800s, sphingolipids remain enigmatic, mostly because of their complexity, structural diversity, and cell specificity. However, increasing attention has been given to sphingolipids in recent years after the discovery of their pivotal role in signal transduction. These molecules are distinguished by the presence of a sphingoid long-chain base (usually sphingosine in mammalian cells) that can be acylated at the 2-amino position to form ceramide (Cer). This simple sphingolipid, a potent signaling molecule by itself, serves as the precursor of more complex sphingolipids such as glycosphingolipids (GSLs) and gangliosides, a subclass of acidic GSLs.
Gangliosides are composed of a common hydrophobic Cer moiety, which acts as a membrane anchor, and a hydrophilic oligosaccharide chain, which varies in length and composition and contains one or more sialic acid residues.1, 2 Most gangliosides are amphipathic constituents of the outer leaflet of cell membranes, where they are vital for the maintenance of membrane structure and organization. A small proportion (10%) is localized in mitochondria and endoplasmic reticulum (ER). The amount and composition of gangliosides in a cell is species- and cell type-specific and may vary dramatically during development and changes in the metabolic state of the cell. Owing to their amphiphilic nature and the unique composition of their hydrophobic portion, gangliosides can either distribute asymmetrically or segregate with cholesterol and other membrane proteins in specialized microdomains or clusters (e.g., lipid rafts and caveolae)3 whose biological activities are greatly influenced by their lipid content.
The diversity and complexity of gangliosides suggest that they are not biologically redundant, but have unique functions as receptors or coreceptors for cytokines, toxins, viruses, and bacteria.1 Gangliosides are also key signaling molecules of pivotal biological processes, including cellular recognition and adhesion, receptor signal transduction, growth regulation, and differentiation.4, 5, 6 In addition, they are important messengers of the adaptive responses to stress such as apoptosis. Under stress conditions that dramatically increase their intracellular concentration, gangliosides can initiate the induction of an apoptotic program.7 Thus, a myriad of crucial cellular responses may be influenced or controlled by gangliosides and ultimately result in either cell growth and division, differentiation, or cell death. Nonetheless, the molecular mechanisms underlying many of these ganglioside-mediated responses remain largely unknown.
Many signal transduction events occur at the plasma membrane and are thought to proceed within caveolae or lipid rafts; these events are greatly influenced by the concentration and subtype of gangliosides.1 Evidence that gangliosides directly perturb membrane composition and permeability or affect the function of membrane components remains circumstantial. However, it is becoming increasingly clear that gangliosides also play crucial roles in subcellular compartments such as the ER and mitochondria.1 At these sites, gangliosides influence often opposite cell fate decisions (e.g., proliferation versus apoptosis), and this action appears to depend on their local concentration, structural characteristics, and sugar modifications.
Underlying many ganglioside-mediated effects is a change in intracellular calcium levels (Ca2+).4, 5 Cytosolic Ca2+ concentration of resting cells is maintained at low levels by the concerted action of specialized channels, a Ca2+ pump, Ca2+-dependent enzymes, and Ca2+-binding proteins. These mechanisms are localized in the cytosol, ER, and mitochondria, the three compartments that control the traffic of Ca2+ across the plasma membrane or into intracellular stores.8 As Ca2+ regulates a plethora of physiological processes, it is not surprising that perturbation of Ca2+ homeostasis is a potent inducer of an ER stress response that, in turn, dictates the fate of the cells.
The scope of this review is to describe the downstream events caused by changes in ganglioside concentration, localization, and composition. These events include activation of an ER stress response, mitochondrial apoptotic signaling, or both that trigger cell death under physiologic or pathologic conditions.
Metabolism of Gangliosides
The biosynthesis of gangliosides takes place in the ER and in the Golgi complex. It is mediated by the action of membrane-bound glycosyltransferases and sialyltransferases, which catalyze the transfer of sugar nucleotide donors to sphingolipid acceptors.1, 9 The first step in ganglioside biosynthesis occurs at the cytosol–ER interface. There, the enzyme UDP-glucose (Glc) Cer glucosyltransferase adds Glc to Cer to form the simplest glycolipid, glucosylceramide (GlcCer). Then, GlcCer translocates across the ER compartment and is rapidly converted into lactosylceramide (LacCer), the common precursor of the GSL series in vertebrates. To form LacCer, galactosyltransferase I transfers a galactose (Gal) moiety from UDP-Gal.9
Complex gangliosides that are more glycosylated are built by the stepwise addition of sugar nucleotides to LacCer. In particular, specific sialyltransferases generate GM3, GD3, and GT3, which are further converted across the cis–medial–trans-Golgi to complex gangliosides of the a-, b-, or c-series, respectively, by sequential addition of Gal, N-acetylgalactosamine (GalNac), or sialic acid residues. In contrast, gangliosides of the 0-series, which include GA1 and GA2, that lack sialic acids are directly formed by LacCer.9 The final products of ganglioside metabolism leave the trans-Golgi network in budding vesicles that have glycan chains oriented toward the extracellular space. These vesicles eventually fuse with the plasma membrane and other intracellular membranes, but maintain their topology9, 10 (Figure 1).

Scheme of the biosynthesis and degradation of gangliosides. Ceramide (Cer) the precursor of all complex sphingolipids is synthesized from sphingosine and fatty acid. The biosynthetic route is identified by black arrows, whereas the degradative route is marked with dark blue arrows. The biosynthesis occurs at the Golgi complex through the action of glycosyltransferases (green), while the degradation occurs in the endosomal/lysosomal compartment by the action of glycosylhydrolases (blue). Both pathways begin and end with ceramide (*). Ganglioside are designated according to the nomenclature of Svennerholm (1980)
The degradation of gangliosides occurs along the endocytic–lysosomal pathway and is controlled by hydrolytic enzymes that function at acidic pH.1 For the efficient catabolism of these membrane-bound substrates, the water-soluble lysosomal hydrolases require the cooperative action of effector proteins named sphingolipid activator proteins (SAPs or saposines). Acting as natural detergents, SAPs facilitate the physical interaction between the enzyme and its substrate.11 Plasma membrane gangliosides are internalized and transported in endocytic vesicles to the lysosomes. After endosome–lysosome fusion, gangliosides expose their glycan chains to the luminal face of the lysosome. This topologic conformation makes gangliosides accessible for degradation by specific exoglycosidases that cleave their sugar residues sequentially from the nonreducing end.1, 2 Initially, a lysosomal sialidase converts multisialogangliosides into monosialogangliosides GM1, GM2, or LacCer. The enzyme β-galactosidase (β-gal) then removes the β-galactosyl moiety from GM1, thereby giving rise to GM2 (Figure 2); and β-N-acetyl-hexosaminidase cleaves the GalNac residue from GM2 to generate GM3. In some cells and species, sialic acid residues are effectively removed from GM1 and GM2 by specific sialidases producing the corresponding asialo derivatives GA1 and GA2. Through the sequential actions of β-gal and β-glucosidase, LacCer is degraded to Cer, and through that of ceramidases, Cer is further converted into sphingosine and fatty acids (Figure 1).

Genetic defect in GM1-gangliosidosis. Lack of β-galactosidase leads to expansion of the lysosomal system and massive accumulation of GM1-ganglioside in neurons of GM1-gangliosidosis mouse model. The presence of numerous vacuoles in the cytoplasm represents the typical histological finding in LSDs (electron microscopy picture)
Recent studies on the metabolic turnover of gangliosides have demonstrated a variety of dynamic processes by which these molecules are modulated. Besides the typical biosynthetic and degradative pathways, specialized glycosidases can modify gangliosides at the plasma membrane; gangliosides can be directly recycled to the plasma membrane from early endosomes;12 they can be sorted to the Golgi apparatus from endosomes and subsequently reglycosylated;12 or they can be fully degraded in lysosomes and reused in the so-called salvage pathway.12 In the latter process, degradative products leave the lysosome and are subsequently modified and reused for the biosynthesis of new GSLs. In some cells, salvage pathways represent an important means of saving energy and can account for as much as 90% of overall ganglioside turnover. The existence of salvage pathways also potentially explains how certain cells cope with a fast turnover of complex gangliosides at the plasma membrane and intracellular membranes during cell division.
Under physiological conditions and at steady-state levels, pools of gangliosides (or ganglioside intermediates) are present in different subcellular compartments. The modulation of their concentration at those sites strictly depends on the coordinated regulation of the biosynthetic, degradative, and salvage/recycling pathways. However, the precise mechanisms by which the cell balances these pathways have not been fully elucidated.
Gangliosides in Disease Pathogenesis
The importance of gangliosides in cellular integrity and homeostasis is made apparent by the many catastrophic pathogenic conditions (e.g., neurodegenerative diseases and cancer) associated with the abnormal expression, degradation, or distribution of these molecules. Regulation of the metabolism of gangliosides in specific cell types is also imperative, as indicated by the numerous human genetic diseases known as GSL storage diseases or glycosphingolipidoses13, 14, 15 (Table 1). These monogenic disorders of metabolism that belong to the large group of the lysosomal storage diseases (LSDs) result from deficiency of any one of the lysosomal enzymes involved in GSL degradation and consequent accumulation of undigested GSLs or their intermediates in lysosomes. An account of the cellular consequences of ganglioside accumulation in lysosomes is given in Figure 2.
Glycosphingolipidoses represent one of the most frequent causes of neurodegeneration and mental retardation in children. These diseases are complex and, in most cases, present with a progressive and severe neurodegenerative course and a broad spectrum of systemic abnormalities.13, 16 The neurologic symptoms include mental retardation or dementia, motor dysfunction, sensory deficits, increased startle response, and seizures.13, 16 In some disorders, cerebellar signs predominate, whereas in others, the cerebral cortex, basal ganglia, or spinal cord neurons are the most affected. The variations in symptoms associated with different glycosphingolipidoses may reflect differences in the metabolic needs of individual cell types that depend on the selective nature of the primary defect. Pathophysiologic studies of patients and animal models have identified changes in neuronal connectivity in the cerebral cortex, including degeneration of axons and synapses of inhibitory neurons (axon swelling or ‘spheroids’), regrowth of dendrites (ectopic dendrites or meganeurites), and formation of new synapses of pyramidal neurons.17, 18, 19, 20 Many of these phenomena have been attributed to ganglioside storage, albeit the underlying molecular effectors are still unknown. Neuronal cell death and demyelination occur in some of these LSDs21, 22, 23 and are often accompanied by astrogliosis and microgliosis that appear mostly in areas of severe neuronal vacuolation.24, 25, 26 The presence of reactive astrocytes is indicative of an elicited neuroinflammatory response, and the biochemical heterogeneity and adaptive plasticity may reflect differences in microenvironmental cues, such as the combination of cytokines, growth factors, adhesion molecules, and other signals emanating from injured neurons, activated microglia, endothelial cells, and vascular components.27, 28, 29
As mentioned in the Introduction, the amount and composition of gangliosides in a cell is species- and cell type-specific. In the central nervous system (CNS), gangliosides constitute 10–12% of the total lipid content, and in the mammalian brain, GM1 ganglioside is the most abundant glycolipid. The ganglioside storage diseases (or gangliosidoses) include the GM1 storage disorder, GM1-gangliosidosis, and the GM2 storage disorders, Tay–Sachs disease, Sandhoff disease, and GM2-gangliosidosis AB variant (Table 1).30, 31 Given the abundance of gangliosides in the CNS, the clinical and pathologic manifestations of these LSDs are characteristic of generalized CNS disorders. Gangliosidoses are severe neurosomatic conditions that occur mainly in infants, although milder forms with later onset and longer survival also occur in adolescents and adults.30, 31 The severe forms are characterized primarily by growth retardation, progressive neurologic deterioration due to extensive brain atrophy, visceromegaly, and skeletal dysplasia.32, 33
In most glycosphingolipidoses, the predominant storage is one particular GSL, for which the corresponding enzyme has the highest affinity (Table 1). Hence, GM1- and GM2-gangliosides accumulate primarily in GM1- and GM2-gangliosidoses. GlcCer and galactosylceramide are the primary storage products in Gaucher disease and Krabbe disease, respectively.13 In some LSDs, gangliosides accumulate secondarily to the primary storage material that may not necessarily be a GSL.34 For example, secondary storage of gangliosides GM2 and GM3 in brain tissue has been reported in the following diseases: Niemann–Pick type A/B disease, in which the primary storage material is sphingomyelin;34 Niemann–Pick type C disease, where the primary storage material is cholesterol;35 and mucopolysaccharidosis types I36 and III,37 in which the primary storage materials are dermatan and heparan sulfate, respectively. As GSLs act as second messengers, their secondary accumulation may be the result of GSL-mediated activation of a signal transduction pathway(s) that controls the synthesis of these molecules. Conceivably, the secondary accumulation of gangliosides may contribute synergistically with the primary storage product to elicit pathogenesis.
In addition to the altered intracellular concentration or distribution of gangliosides that occur as a consequence of lysosomal storage, the presence of free pools of gangliosides in the blood plasma, cerebrospinal fluid (CSF), and other body fluids could be part of the molecular mechanisms of disease. Under physiologic conditions, a constant exchange appears to occur between cell-associated and non-cell-associated gangliosides.38 However, increased turnover of cell membranes resulting from cell degeneration or cell growth leads to an increased release of gangliosides into the extracellular milieu by a process called ‘shedding’.39
Several studies have suggested that gangliosides in the CSF are shed from plasma membranes of neural cells. Degenerative processes in the CNS have been linked to increased shedding of membrane fragments into the intercellular space.5, 40, 41 For example, ganglioside GD3, which is present only in trace amounts in the normal adult brain, is expressed at high levels in activated microglia and in reactive astrocytes.40, 42 GD3 is released by primary murine microglial cells under neuroinflammatory conditions and is directly responsible for the induction of apoptosis in oligodendrocytes43 (GD3-mediated apoptotic response is discussed in detail below). Increased GD3 expression has also been detected in brain tissue from patients with various neurodegenerative disorders such as Creutzfeld–Jacob disease and multiple sclerosis.44, 45 Moreover, elevated CSF levels of GM3 and to a lesser extent GD3 have been associated with pronounced dysfunction of the blood–brain barrier.46 On the basis of these observations, we predict that altered intracellular expression of GM3 and GD3 and their release into the extracellular milieu occur in other neurodegenerative and neuroinflammatory conditions including some LSDs. We also speculate that these gangliosides actively participate in cell degeneration.
Animal Models of Gangliosidoses
By elucidating the complex underlying mechanisms of disease pathogenesis linked to the accumulation of gangliosides, we will improve our knowledge of the physiologic functions of these molecules.7, 47 Progress toward this endeavor has been made by generating mice with targeted disruptions of key genes that encode glycosyltransferases or glycosylhydrolases, the enzymes that control the synthesis and degradation of gangliosides.48, 49, 50 Although no human diseases have yet been identified in which pathogenesis is attributed to genetic deficiency of any glycosyltransferase, the analyses of these genetically engineered null mice are beginning to identify the functions of complex gangliosides in development and cell differentiation. For instance, deficiency of glucosylceramide synthase gene (Ugcg), which eliminates the major pathway of ganglioside synthesis,51 does not adversely affect growth rate, and differentiation in vitro, but impairs embryonic development and differentiation of some tissues in vivo.51 In contrast, β-1,4-N-acetylgalactosaminyltransferase (GalNAcT)-knockout mice express predominantly GM3- and GD3-gangliosides in their CNS, but have virtually normal CNS development and a normal lifespan.52 The outcome of these studies can be explained, at least in part, by a possible compensatory mechanism in which precursors of complex gangliosides take over some of the functions attributed to the end products. Considerably more severe phenotypes have been demonstrated in mice carrying mutations in two crucial ganglioside-specific glycosyltransferase genes, Siat9, that encodes GM3 synthase (CMP-N-acetylneuraminic acid : LacCer α-2,3-sialyltransferase) and Galgt1 that encodes GM2/GD2 synthase (UDP-N-acetyl-D-galactosamine : GM3/GM2/GD2 synthase), also known as GalNAcT.50 These mice are unable to synthesize gangliosides of the a-, b-, or c-series. Soon after weaning, viable mice develop a severe neurodegenerative disease that results in premature death. Histopathologic analyses of the CNS revealed striking vacuolization of the white matter regions, axonal degeneration, and perturbed axon–glia interactions.50 Overall, these studies underscore the crucial role of gangliosides in stabilizing the developing CNS.
Spontaneous genetic defects in the lysosomal enzymes that degrade GSLs have been identified in various mammalian species53 and used primarily for comparative assessments of pathologic abnormalities and for the implementation of therapeutic modalities. Although valuable, these large animal models are difficult to handle and maintain as colonies for scientific purposes. However, the generation of mouse models of these genetic diseases by gene targeting in embryonic stem (ES) cells have revolutionized the field and allowed for in-depth, comprehensive studies that were either impossible or extremely laborious to pursue in large animals or patients.
Most of the LSDs occurring in man have now been reproduced in the mouse.54 Despite slight phenotypic variations caused by differences in metabolic pathways of the two species55 and the influence of genetic background, mouse models of glycosphingolipidoses appear to closely resemble the corresponding human diseases (Table 1).48, 49, 56 For instance, GM1-gangliosidosis mice develop a generalized nervous system condition characterized by tremors, ataxia, and abnormal gait that culminates in rigidity and paralysis of the hind limbs and premature death.48, 56 However, unlike the human disorders, the two GM2-gangliosidosis mouse models show different neurologic phenotypes. Although they display biochemical and pathologic features of the disease, the Tay–Sachs mice are asymptomatic early in life and develop chronic neurologic abnormalities.57 In contrast, Sandhoff mice present with a severe and rapidly progressive neurodegenerative course.58 In the CNS of Tay–Sachs and GM1 mice, a neuraminidase efficiently desialylates the GM1 and GM2 into their corresponding asialo derivatives, GA1 and GA2, that accumulate in massive amounts as the animals age.48, 57 In the GM1 mouse model, this phenotype is accompanied by gradual deterioration of motor functions48, 56 and progressive CNS inflammation.28 Owing to their similarities to the corresponding human diseases, these mouse models are a proverbial gold mine for studying molecular pathogenesis associated with altered ganglioside metabolism.
Gangliosides and the ER Stress Response
The presence of gangliosides at the ER membrane implies that these molecules regulate membrane dynamics and structure in this organelle; hence, changes in ganglioside composition, sugar modifications, and local concentration can alter not only their behavior but also that of other membrane components. This action can have deleterious consequences. It has been postulated that gangliosides at the ER and nuclear membranes act as key modulators of Ca2+ flux,4 although the molecular mechanisms underlying this function are not fully elucidated. The ER is the primary store of intracellular Ca2+, especially in neurons. The Ca2+ concentration of the ER is maintained around 5 mM, and that of the cytosol is kept as low as 0.1 μM59 by the synchronous action of membrane-bound and -soluble Ca2+-binding proteins localized at different subcellular sites, including the ER. This organelle is, therefore, directly responsible for controlling cellular processes mediated by Ca2+ signaling (e.g., post-translational protein folding and maturation). Several ER-resident proteins involved in the folding process require Ca2+ binding for their activity.60, 61 Thus, depletion of the ER Ca2+ store interferes with the folding pathway, results in the accumulation of misfolded protein aggregates, and can potentially damage cells. To prevent this problem and ensure the release of correctly folded proteins from the ER, eukaryotic cells have developed a quality control system called the ‘ER stress response’ or ‘unfolded protein response’ (UPR) that monitors the physiologic state of the ER.62
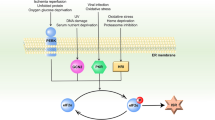
The ER stress response promotes cell survival under stress conditions by using mechanisms such as transcriptional upregulation of ER-resident chaperones and folding catalysts (e.g., BiP and PDI) that depend on an adequate Ca2+ concentration within the ER lumen, translation attenuation, and degradation. Together, these responses limit the further accumulation of unfolded proteins, prevent the aggregation of existing ones, and dispose of terminally unfolded proteins.62 However, the irreversible damage caused by prolonged ER stress triggers an apoptotic program that ultimately leads to cell death (Figure 3) (see reviews in this issue).

Proapoptogenic interorganellar crosstalk triggered by gangliosides. GM1 accumulation at the ER membrane induces release of Ca2+ to the cytosol. Depletion of ER-Ca2+ store induces activation of an ER stress response accompanied by the activation, among others, of BiP, ATF6, CHOP, JNK2, PERK and caspase-12. Cytosolic Ca2+ is rapidly taken up by mitochondria and its increase in the mitochondrial matrix induces MMP with release of apoptogenic factors such as cytochrome c and Smac/Diablo. The formation of the apoptosome with the activation of caspase-3 leads to apoptosis. Ganglioside GD3 acts directly on the outer mitochondrial membrane through opening of the PTP. However, the direct effect of gangliosides on the Bcl-2 proapoptogenic proteins is unknown
Under normal cellular conditions, the ER-resident chaperone BiP associates with the luminal portions of the following three ER-resident UPR sensors located at the ER membrane: the Ser/Thr kinase and ribonuclease IRE1, the basic Leu-zipper transcription factor ATF6, and the Ser/Thr kinase PERK63 (Figure 3). Once unfolded or misfolded proteins begin to accumulate in the ER, they preferentially recruit BiP away from the UPR sensors. Consequently, PERK and IRE1 undergo homodimerization, transautophosphorylation, and activation; ATF6 is transported to the Golgi, where limited proteolysis converts it into an active transcription factor.63 Processed ATF6 can then translocate into the nucleus, where it activates the transcription of genes that contain an ER stress-responsive element in their promoter. These include chaperones like BiP and calreticulin, which aid in the folding process, and the proapoptotic transcription factor CHOP/GADD153, which decreases the expression of antiapoptotic Bcl-2 and can therefore induce apoptosis.64
Upon ER stress-mediated oligomerization, IRE1 is proteolytically cleaved into soluble C-terminal fragments with both kinase and ribonuclease activity. Similarly to ATF6, the IRE1 fragments enter the nucleus and upregulate ER stress-responsive genes.65 The IRE-mediated response is, however, slower than that mediated by ATF6, because it occurs via processing the mRNA for XBP1, which is induced by active ATF6. Splicing of a small intron from the XBP1 mRNA is carried out by the ribonuclease activity of IRE1 and generates an active transcription factor. Activated IRE1 can also recruit the cytosolic adaptor protein TRAF2 (tumor necrosis factor receptor–associated factor 2), which in turn binds and activates JNK and initiates a proapototic signal.66 Thus, cellular responses to ER stress involve molecules that modulate the transcription of folding catalysts, chaperones, or proapoptotic factors, the choice of which most likely depends on the level, extent, and type of stressor. On the other hand, ER stress-activated PERK attenuates translation.67
PERK phosphorylates the α subunit of the eukaryotic translation initiation factor 2 (eIF2) that prevents the assembly of the 80S ribosome and consequently inhibits protein synthesis. This response is very rapid, because eIF2 is the direct target of PERK. Therefore, PERK functions as both the sensor and effector of ER stress. This theory is supported by the observation that PERK−/− murine ES cells lose the ability to phosphorylate eIF2α and to attenuate translation upon accumulation of unfolded proteins.67 It remains to be determined how ER chaperones escape from PERK-mediated inhibition of translation initiation and are induced to cope with unfolded proteins.
When cells are exposed to excess levels of stimuli that cause ER stress, the apoptotic program supersedes the survival program by mechanisms that are not fully understood. The concentration of Ca2+ not only in the ER but also in the mitochondria and cytosol might represent an additional cause of apoptosis. A key player in this process is the cysteine protease caspase-12 whose transcription is induced during an ER stress response.7, 68, 69 It has been postulated that caspase-12 contributes to cell death in ischemic brain and other neurodegenerative conditions, including Hungtington and Alzheimer disease.69 Although the molecular pathways that link ER stress to caspase-12 induction are not fully elucidated, caspase-12−/− cortical neurons are resistant to amyloid β-protein-mediated neurotoxicity (Figure 3).69
Under normal conditions, procaspase-12 is localized at the cytoplasmic side of the ER membrane in a complex with other molecules such as unphosphorylated TRAF2, which transduces signals from IRE1.70 When the cytosolic concentration of Ca2+ becomes toxic, an apoptotic cascade initiates with the oligomerization and proteolytic activation of procaspase-12 by the Ca2+-dependent protease m-calpain.59, 69 Caspase-12 also appears to be able to undergo self-maturation, acting directly on procaspase-12.71 Proteolytic activation of caspase-12 results in a caspase cascade that involves the effector caspases, caspase-9 and caspase-3.71 After caspase-12 is activated, TRAF2 dissociates from the complex, which in turn initiates both IRE1- and JNK-mediated cell death pathways.70 JNK participates in the release of cytochrome c from mitochondria after TRAF2 induction through a Bim/Bax-mediated mechanism.72 It is now evident, however, that although caspase-12 appears to be the principal effector of apoptosis, other pathways are involved in ER stress-induced cell death signals. For instance, caspase-8 appears to act independently of caspase-12 and mediates the release of mitochondrial cytochrome c via activation of Bid/Bax pathway and the cleavage of the BiP-associated protein BAP31, which is a transmembrane ER protein.73
Like the caspase-8 pathway, many other biochemical pathways are known to be activated either by depletion of ER Ca2+ stores or by elevation of cytosolic Ca2+ levels. For instance, in the nervous system, the increased frequency and magnitude of Ca2+ transients results in increased growth rates of neuronal processes, yet gradual depletion of ER Ca2+ results in failure to generate Ca2+ transients that are essential for neurite outgrowth (neuritogenesis).74 It has been proposed that activation of one or more of these pathways by Ca2+ imbalance is a downstream response to GSL accumulation. The mechanisms that regulate cytosolic Ca2+ concentration entail external Ca2+ influx via voltage- and ligand-gated channels at the plasma membrane and the release of Ca2+ from intracellular stores, that is, ER and mitochondria. In the ER lumen, Ca2+ is either free or bound to luminal Ca2+-binding proteins such as calnexin and calreticulin. Efflux of Ca2+ from the ER occurs via two types of channels that reside in the ER membrane, the Ca2+-gated Ca2+-release channel or ryanodine receptor (RyaR) and the inositol 1,4,5-triphosphate-gated Ca2+-release channel, the IP3 receptor (IP3R). Ca2+ influx from the cytosol to the ER lumen is mediated by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA).75, 76 Fluctuations in Ca2+ concentration help regulate normal cell functions,8 but ER Ca2+ imbalance and acute Ca2+ release from the ER lead to cell death. The aforementioned pump and channels at the ER membrane may, at least in part, control both of these conditions.
There is increasing evidence that these regulators of the Ca2+ concentration in the ER can be altered directly or indirectly by the level and type of GSLs, including gangliosides, that are present at the ER membrane.47, 77, 78 For instance, accumulation of GlcCer, which is the main storage product in Gaucher disease, increases calcium mobilization from intracellular stores in cultured neurons, likely via amplification of the response of the RyaR to agonists, which leads to ER calcium release.47, 77 As a result, neurons show increased sensitivity to neurotoxic agents and Ca2+-mediated cell death. Isolated microsomes from the brain of a Sandhoff disease mouse model that accumulate GM2 show substantially reduced rate of Ca2+ uptake because the Vmax of SERCA is reduced in those organelles.47 This effect of GM2 is prevented if the mice are treated with a specific inhibitor of glycolipid synthesis that reduces GM2 storage, directly implicating this ganglioside in the function of SERCA.47 Thus, neuronal Ca2+ homeostasis is perturbed in Gaucher and Sandhoff diseases resulting in elevated cytosolic Ca2+ levels, which may lead to enhanced sensitivity of the neurons to stress and promote cell death.
The molecular mechanisms by which GM2 modulates SERCA, and GlcCer modulates the RyaR have not been elucidated. However, recent studies have helped to add a piece to the puzzle that could eventually link the effects of ganglioside accumulation on the Ca2+ homeostasis and neurodegeneration.7 Tessitore et al.7 demonstrated that in neurons of the GM1-gangliosidosis mouse model, excessive accumulation of GM1 in lysosomes results in the build-up of this ganglioside at the ER membrane, which in turn depletes ER Ca2+ stores and activates the ER stress response. The combined upregulation of BiP and CHOP increases the levels of processed ATF6 and activates JNK2 and caspase-12, which ultimately leads to neuronal apoptosis. The ER-stress-mediated apoptotic pathway is not induced in β-gal−/−/GalNAcT−/−, which are deficient for β-gal and GM2-ganglioside synthase and do not accumulate GM1; this finding directly implicates GM1 in the activation of the apoptotic pathway.
Increased GM1 concentration at the ER may trigger the ER stress response either by directly affecting Ca2+ transport across the membrane or by indirectly influencing the activity of other membrane components, including the Ca2+ pump and channels. The latter possibility would be consistent with the observed effect of GM2 accumulation on the activity of SERCA in microsomal membranes from Sandhoff disease mice and the cholesterol-induced ER stress response in macrophages from Niemann–Pick A/B mice.47, 79 Ultimately, the disruption of intracellular Ca2+ homeostasis in ganglioside-accumulating cells could affect protein folding in the ER and induce the ER stress response through the conventional route, thereby suggesting a novel mechanism of neuronal apoptosis that could also occur in other neurodegenerative diseases.7
Gangliosides as Propagators of the ER Stress- and Mitochondria-Elicited Death Pathways
Accumulating evidence suggests that different intracellular organelles contribute synergistically to the initiation of apoptosis by specific stress inducers. Therefore, apoptotic signals emanating from one subcellular compartment relay to the rest of the cell. The interplay between the ER and the mitochondrion in controlling intracellular Ca2+ concentration directly affects the apoptotic signaling network; in turn, the influence of gangliosides on the membrane topology and function of these two organelles may alter their cellular response to stressors.
Experimental evidence argues for a role of GM1 in ER stress response-induced Ca2+ release from the ER when GM1 concentration at this site reaches a certain threshold. One means of maintaining Ca2+ homeostasis is by the rapid uptake of cytosolic Ca2+ into the mitochondria, which depends on a negative transmembrane potential and a Ca2+ uniporter.80 Thus, GM1 accumulation in the ER may simultaneously or secondarily affect the function of the closely juxtaposed mitochondria by provoking mitochondrial Ca2+ overload.81, 82
Most of the downstream effects of Ca2+ release from the ER hinge on the disruption of the mitochondrial membrane permeability (MMP) due to the opening of the permeability transition pore (PTP).83 The PTP is a high-conductance, nonselective megachannel that forms by the apposition of transmembrane proteins from the inner and outer mitochondrial membranes.82 Opening of the PTP can be triggered by several physiologic and pathologic processes that occur simultaneously or consequentially; these include increase in Ca2+ levels in the mitochondrial matrix, production of reactive oxygen species and nitric oxide, changes in pH, lowering of the mitochondrial transmembrane potential, and increase in the concentration of GD3.82, 84 PTP opening causes swelling and rupture of the mitochondria with a sudden increase in the permeability of the inner mitochondrial membrane. This so-called ‘permeability transition’ (PT) results in immediate dissipation of the proton-dependent mitochondrial transmembrane potential and chemical equilibration between the cytoplasm and mitochondrial matrix. It also allows the release of diffusible apoptogenic factors, which are normally confined between the inner and outer mitochondrial membranes; these include cytochrome c, apoptosis-inducible factor (AIF), and second mitochondrial activator of caspases (SMAC/Diablo).82, 85 Cytosolic cytochrome c induces the formation of the apoptosome and activates caspases and nucleases that finalize the apoptotic process (Figure 3).
Cytochrome c released from mitochondria binds directly and selectively to IP3R; thus, cytosolic access of cytochrome c is restricted early in apoptosis. Also, cytochrome c binding to IP3R abolishes inhibition of IP3R-associated Ca2+ release. By blocking the feedback regulation of IP3R, cytochrome c greatly augments the release of Ca2+, which then is rapidly taken up by juxtaposed mitochondria to provoke further cytochrome c release. This feedforward amplification manifested by sustained increases in cytosolic Ca2+ during early apoptosis elicits massive release of cytochrome c from all mitochondria.86 For instance, in neurons, cytochrome c release at a focal point within the cell such as the cell body or neurites can create a wave of cytochrome c release throughout the cell body and cause further damage.
Agents capable of inducing mitochondrial PT and consequent release of cytochrome c are considered important mediators of apoptotic signals, and GD3, which directly targets mitochondria by opening the PTP, can be listed among the most potent natural inducers.5, 87 Interestingly, Scorrano et al.84 have demonstrated that GD3 preferentially affect mitochondrial function, while other gangliosides such as GD1a, GM3 have no effect. Acetylation of GD3 nullifies its apoptotic effect and this mechanism is suggested to confer resistance to tumors.88 These findings imply that differences in ganglioside chemical structures modify their functions. Although Ca2+ appears to be unnecessary in GD3-mediated PT, elevated intracellular Ca2+ can synergistically accelerate the induction of this event. Ca2+ also regulates the association between mitochondria and a smooth domain of the ER and enhances the ability of gangliosides to produce membrane clusters.89 Whether mitochondrial damage occurs through ganglioside interaction with a candidate component of the PTP or through direct perturbation of mitochondrial membranes and, in turn, PTP function, remains to be elucidated. However, the fact that GD3-mediated opening of PTPs is antagonized by known PT inhibitors (e.g., cyclosporin A, ADP, trifluoperazine, and Mg2+) suggests that GD3 acts directly at the pore level. Additionally, overexpression of calnexin, an ER chaperone, retains GD3 synthase in the ER compartment and, in turn, suppresses its apoptotic activity by preventing GD3 to reach the mitochondria.90
Other potential downstream targets of gangliosides are the Bcl-2 family of proteins. Pro- and antiapoptotic Bcl-2 family members are involved in the ER stress response-mediated apoptotic pathway. These proteins, which were thought to act exclusively in the mitochondria, are also localized in the ER.91 Hence, they are present in key locations where Ca2+ is transported, and they may help control Ca2+ flux. Antiapoptotic Bcl-2, a small integral membrane protein, localizes to the ER membrane where it is thought to decrease the Ca2+ content of the ER and reduce the Ca2+ efflux via the IP3R to the mitochondria.92 On the other hand, increased levels of Bcl-2 at the ER enhance the Ca2+ efflux by physical interaction of Bcl-2 with IP3R-1.93, 94 Although the molecular mechanism underlying this phenomenon is still unclear, these data suggest a Ca2+-mediated proapoptotic function of Bcl-2 in the ER that probably depends on its local concentration and interaction with other membrane components, whose function may be influenced by gangliosides.95
The ER-localized proapoptotic Bax and Bak undergo conformational changes and oligomerization upon Ca2+-induced ER stress.96 In turn, the proteins promote proteolytic processing of the ER-resident caspase-12 leading to cell death. In fact, targeted overexpression of Bak in the ER depletes its Ca2+ levels, activates caspase-12, and ultimately causes cell death. Thus, the ratio of Bax and Bak to Bcl-2 appears to be a key determinant of the levels of Ca2+ in the ER, as inferred from earlier studies.93, 97 In cells lacking Bax and Bak, uninhibited Bcl-2 leads to IP3R-1 hyperphosphorylation, enhanced ER Ca2+ release, and decreased steady-state ER Ca2+ stores.94 Finally, Bid, Bik, and Bim, the upstream regulators of these Bcl-2 family members, are also influenced by proximal signals including ER stress. Considering that gangliosides and some of the Bcl-2 proteins colocalize at intracellular membranes, it is conceivable that changes in ganglioside concentration may activate signal pathways attributed to Bcl-2 family of proapoptotic proteins. Hence, controlling the intracellular levels of gangliosides appears to be crucial for the cell to determine its fate.
Conclusion
The finding that gangliosides are active mediators of apoptotic programs has opened a remarkable and interesting field of research that has improved our knowledge of the function of these molecules in physiologic and pathologic conditions. Understanding the hierarchy in interorganelle crosstalk and the way that ER and mitochondria interact in response to stress will help to unravel the multifaceted roles of gangliosides in the apoptosis and signal transduction pathways. So far, Ca2+ signaling appears to be the key player in the apoptotic arena, but the molecular mechanism(s) by which gangliosides perturb intracellular Ca2+ flux that ultimately result in cell death need to be further clarified. The Bcl-2 family of proteins could serve as intermediate components of the ganglioside-mediated apoptotic program induced by disturbance of the ER Ca2+ homeostasis. This hypothesis does not necessarily exclude the possibility that gangliosides may also directly affect the MMP and cause mitochondrial damage. In either of the scenarios, gangliosides integrate a plethora of cellular responses by impinging on the functional integrity of intracellular membranes. A full understanding of the involvement of gangliosides in the cell death program will allow us to target molecular effectors that would potentially prevent apoptosis-modulating signals in numerous conditions where gangliosides appear to be harmful.
Abbreviations
- Glc:
-
glucose
- Gal:
-
galactose
- GalNac:
-
N-acetylgalactosamine
- Lac:
-
lactose
- Cer:
-
ceramide
- ER:
-
endoplasmic reticulum
- GSLs:
-
glycosphingolipids
- Ca2+:
-
calcium
- GlcCer:
-
glucosylceramide
- LacCer:
-
lactosylceramide
- SAPs:
-
saposines
- LSDs:
-
lysosomal storage diseases
- CSF:
-
cerebrospinal fluid
- CNS:
-
central nervous system
- ES:
-
embryonic stem
- UPR:
-
unfolded protein response
- SERCA:
-
sarco/endoplasmic reticulum Ca2+-ATPase
- PTP:
-
permeability transition pore
- MMP:
-
mitochondrial membrane permeabilization
- GalNAcT:
-
β-1,4-N-acetylgalactosaminyltransferase
- RyaR:
-
ryanodine receptor
- IP3R:
-
IP3 receptor
- SMAC/Diablo:
-
second mitochondrial activator of caspases
References
Huwiler A, Kolter T, Pfeilschifter J and Sandhoff K (2000) Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. Acta 1485: 63–99
Tettamanti G (2004) Ganglioside/glycosphingolipid turnover: new concepts. Glycoconjugate J. 20: 301–317
Heffer-Lauc M, Lauc G, Nimrichter L, Fromholt SE and Schnaar RL (2005) Membrane redistribution of gangliosides and glycosylphosphatidylinositol-anchored proteins in brain tissue sections under conditions of lipid raft isolation. Biochim. Biophys. Acta 1686: 200–208
Ledeen RW and Wu G (2002) Ganglioside function in calcium homeostasis and signaling. Neurochem. Res. 27: 637–647
Malisan F and Testi R (2002) GD3 ganglioside and apoptosis. Biochim. Biophys. Acta 1585: 179–187
Miljan EA and Bremer EG (2002) Regulation of growth factor receptors by gangliosides. Sci. STKE 2002: RE15
Tessitore A, del PMM, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM and d'Azzo A (2004) GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell 15: 753–766
Berridge MJ, Bootman MD and Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 4: 517–529
Kolter T, Proia RL and Sandhoff K (2002) Combinatorial ganglioside biosynthesis. J. Biol. Chem. 277: 25859–25862
Yu RK, Bieberich E, Xia T and Zeng G (2004) Regulation of ganglioside biosynthesis in the nervous system. J. Lipid Res. 45: 783–793
Kolter T and Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell. Dev. Biol. 21: 81–103
Tettamanti G, Bassi R, Viani P and Riboni L (2003) Salvage pathways in glycosphingolipid metabolism. Biochimie 85: 423–437
Scriver C, Beaudet A, Sly W and Valle D (eds) (2001) The Metabolic & Molecular Bases of Inherited Disease (New York: McGraw-Hill)
D'Azzo A (2003) Gene transfer strategies for correction of lysosomal storage disorders. Acta Haematol. 110: 71–85
Futerman AH and van Meer G (2004) The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell. Biol. 5: 554–565
Ginzburg L, Kacher Y and Futerman AH (2004) The pathogenesis of glycosphingolipid storage disorders. Semin. Cell Dev. Biol. 15: 417–431
Walkley SU, Zervas M and Wiseman S (2000) Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb. Cortex 10: 1028–1037
Walkley SU (1998) Cellular pathology of lysosomal storage disorders. Brain Pathol. 8: 175–193
Jolly RD and Walkley SU (1997) Lysosomal storage diseases of animals: an essay in comparative pathology. Vet. Pathol. 34: 527–548
Jeyakumar M, Dwek RA, Butters TD and Platt FM (2005) Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 6: 713–725
Kaye EM, Alroy J, Raghavan SS, Schwarting GA, Adelman LS, Runge V, Gelblum D, Thalhammer JG and Zuniga G (1992) Dysmyelinogenesis in animal model of GM1 gangliosidosis. Pediatr. Neurol. 8: 255–261
Wenger DA (2000) Murine, canine and non-human primate models of Krabbe disease. Mol. Med. Today 6: 449–451
Zaka M and Wenger DA (2004) Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci. Lett. 358: 205–209
German DC, Liang CL, Song T, Yazdani U, Xie C and Dietschy JM (2002) Neurodegeneration in the Niemann–Pick C mouse: glial involvement. Neuroscience 109: 437–450
Raivich G, Bohatschek M, Kloss C, Werner A, Jones L and Kreutzberg G (1999) Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res. Rev. 30: 77–105
Wada R, Tifft CJ and Proia RL (2000) Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 97: 10954–10959
Suzuki K (2003) Globoid cell leukodystrophy (Krabbe's disease): update. J. Child. Neurol. 18: 595–603
Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d'Azzo A, Perry VH, Butters TD, Dwek RA and Platt FM (2003) Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126: 974–987
Sano R, Tessitore A, Ingrassia A and d'Azzo A (2005) Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood 106: 2259–2268
Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K and Suzuki K (2001) The GM2 Gangliosidoses In The Metabolic & Molecular Bases of Inherited Disease, Scriver C, Beaudet A, Sly W, Valle D (eds) (New York: McGraw-Hill Publishing) pp 3827–3876
Suzuki Y, Oshima A and Nanba E (2001) b-galactosidase deficiency (b-galactosidosis): GM1 gangliosidosis and Morquio B disease In The Metabolic and Molecular Bases of Inherited Disease, Scriver C, Beaudet A, Sly W, Valle D (eds) (New York: McGraw-Hill Publishing Co.) pp 3775–3809
Georgiou T, Drousiotou A, Campos Y, Caciotti A, Sztriha L, Gururaj A, Ozand P, Zammarchi E, Morrone A and D'Azzo A (2004) Four novel mutations in patients from the Middle East with the infantile form of GM1-gangliosidosis. Hum. Mutat. 24: 352
van der Voorn JP, Kamphorst W, van der Knaap MS and Powers JM (2004) The leukoencephalopathy of infantile GM1 gangliosidosis: oligodendrocytic loss and axonal dysfunction. Acta Neuropathol. (Berlin) 107: 539–545
Walkley SU (2004) Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 15: 433–444
Ikonen E and Holtta-Vuori M (2004) Cellular pathology of Niemann–Pick type C disease. Semin. Cell Dev. Biol. 15: 445–454
Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY and Clarke LA (1998) Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin. Genet. 53: 349–361
Jones MZ, Alroy J, Rutledge JC, Taylor JW, Alvord Jr EC, Toone J, Applegarth D, Hopwood JJ, Skutelsky E, Ianelli C, Thorley-Lawson D, Mitchell-Herpolsheimer C, Arias A, Sharp P, Evans W, Sillence D and Cavanagh KT (1997) Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 56: 1158–1167
Ziegler-Heitbrock HW, Kafferlein E, Haas JG, Meyer N, Strobel M, Weber C and Flieger D (1992) Gangliosides suppress tumor necrosis factor production in human monocytes. J. Immunol. 148: 1753–1758
Tsuboi N, Utsunomiya Y, Kawamura T, Kikuchi T, Hosoya T, Ohno T and Yamada H (2003) Shedding of growth-suppressive gangliosides from glomerular mesangial cells undergoing apoptosis. Kidney Int. 63: 936–946
Andersson LM, Fredman P, Lekman A, Rosengren L and Gisslen M (1998) Increased cerebrospinal fluid ganglioside GD3 concentrations as a marker of microglial activation in HIV type 1 infection. AIDS Res. Hum. Retroviruses 14: 1065–1069
Trbojevic-Cepe M, Kracun I, Jusic A and Pavlicek I (1991) Gangliosides of human cerebrospinal fluid in various neurologic diseases. J. Neurol. Sci. 105: 192–199
Kawai K, Kuroda S, Watarai S, Takahashi H and Ikuta F (1994) Occurrence of GD3 ganglioside in reactive astrocytes – an immunocytochemical study in the rat brain. Neurosci. Lett. 174: 225–227
Simon BM, Malisan F, Testi R, Nicotera P and Leist M (2002) Disialoganglioside GD3 is released by microglia and induces oligodendrocyte apoptosis. Cell Death Differ. 9: 758–767
Ohtani Y, Tamai Y, Ohnuki Y and Miura S (1996) Ganglioside alterations in the central and peripheral nervous systems of patients with Creutzfeldt–Jakob disease. Neurodegeneration 5: 331–338
Yu RK, Ledeen RW and Eng LF (1974) Ganglioside abnormalities in multiple sclerosis. J. Neurochem. 23: 169–174
Bektas M and Spiegel S (2004) Glycosphingolipids and cell death. Glycoconjugate J. 20: 39–47
Pelled D, Lloyd-Evans E, Riebeling C, Jeyakumar M, Platt FM and Futerman AH (2003) Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 278: 29496–29501
Hahn C, Martin M, Schröder M, Vanier M, Hara Y, Suzuki K, Suzuki K and d'Azzo A (1997) Generalized CNS disease and massive Gm1-ganglioside accumulation in mice defective in lysosomal acid β-galactosidase. Hum. Mol. Genet. 6: 205–211
Proia RL (2001) Cloning the beta-hexosaminidase genes. Adv. Genet. 44: 127–135
Yamashita T, Wu YP, Sandhoff R, Werth N, Mizukami H, Ellis JM, Dupree JL, Geyer R, Sandhoff K and Proia RL (2005) Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon–glial interactions. Proc. Natl. Acad. Sci. USA 102: 2725–2730
Yamashita T, Wada R, Sasaki T, Deng C, Bierfreund U, Sandhoff K and Proia R (1999) A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. USA 96: 9142–9147
Liu Y, Wada R, Kawai H, Sango K, Deng C, Tai T, McDonald MP, Araujo K, Crawley JN, Bierfreund U, Sandhoff K, Suzuki K and Proia RL (1999) A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. J. Clin. Invest. 103: 497–505
Craigen WJ (2001) Mouse models of human genetic disorders In The Metabolic & Molecular Bases of Inherited Disease, Scriver C, Beaudet A, Sly W, Valle D (eds) (New York: McGraw-Hill Publishing Co.) pp 379–415
Suzuki K, Proia R and Suzuki K (1998) Mouse models of human lysosomal diseases. Brain Pathol. 8: 195–215
Elsea SH and Lucas RE (2002) The mousetrap: what we can learn when the mouse model does not mimic the human disease. Ilar. J. 43: 66–79
Itoh M, Matsuda J, Suzuki O, Ogura A, Oshima A, Tai T, Suzuki Y and Takashima S (2001) Development of lysosomal storage in mice with targeted disruption of the beta-galactosidase gene: a model of human G(M1)-gangliosidosis. Brain Dev. 23: 379–384
Yamanaka S, Johnson MD, Grinberg A, Westhphal H, Crawley JN, Taniike M, Suzuki K and Proia RL (1994) Targeted disruption of the Hexa gene results in mice with biochemical and pathologic features of Tay–Sachs disease. Proc. Natl. Acad. Sci. USA 91: 9975–9979
Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K and Proia RL (1995) Mouse models of Tay–Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 11: 170–176
Orrenius S, Ankarcrona M and Nicotera P (1996) Mechanisms of calcium-related cell death. Adv. Neurol. 71: 137–149 (discussion 149–151)
Back SH, Schroder M, Lee K, Zhang K and Kaufman RJ (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35: 395–416
Bonilla M, Nastase KK and Cunningham KW (2002) Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 21: 2343–2353
Schroder M and Kaufman RJ (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74: 739–789
Patil C and Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13: 349–355
McCullough KD, Martindale JL, Klotz LO, Aw TY and Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21: 1249–1259
Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H and Ron D (1998) Identification of novel stress-induced genes downstream of chop. EMBO J. 17: 3619–3630
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP and Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666
Harding HP, Zhang Y, Bertolotti A, Zeng H and Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5: 897–904
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA and Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103
Nakagawa T and Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150: 887–894
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T and Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276: 13935–13940
Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE and Ellerby HM (2002) Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J. Biol. Chem. 277: 21836–21842
Lei K and Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 100: 2432–2437
Jimbo A, Fujita E, Kouroku Y, Ohnishi J, Inohara N, Kuida K, Sakamaki K, Yonehara S and Momoi T (2003) ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp. Cell Res. 283: 156–166
Wu G, Lu ZH and Ledeen RW (1995) Induced and spontaneous neuritogenesis are associated with enhanced expression of ganglioside GM1 in the nuclear membrane. J. Neurosci. 15: 3739–3746
Brini M, Bano D, Manni S, Rizzuto R and Carafoli E (2000) Effects of PMCA and SERCA pump overexpression on the kinetics of cell Ca(2+) signalling. EMBO J. 19: 4926–4935
Szabadkai G, Simoni AM and Rizzuto R (2003) Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278: 15153–15161
Lloyd-Evans E, Pelled D, Riebeling C, Bodennec J, de-Morgan A, Waller H, Schiffmann R and Futerman AH (2003) Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 278: 23594–23599
Wang Y, Tsui Z and Yang F (1999) Antagonistic effect of ganglioside GM1 and GM3 on the activity and conformation of sarcoplasmic reticulum Ca(2+)-ATPase. FEBS Lett. 457: 144–148
Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D and Tabas I (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5: 781–792
Babcock DF and Hille B (1998) Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 8: 398–404
Boya P, Cohen I, Zamzami N, Vieira HL and Kroemer G (2002) Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 9: 465–467
Ferri KF and Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3: E255–E263
Green DR and Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629
Scorrano L, Petronilli V, Di Lisa F and Bernardi P (1999) Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J. Biol. Chem. 274: 22581–22585
Hanson CJ, Bootman MD and Roderick HL (2004) Cell signalling: IP3 receptors channel calcium into cell death. Curr. Biol. 14: R933–R935
Boehning D, van Rossum DB, Patterson RL and Snyder SH (2005) A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc. Natl. Acad. Sci. USA 102: 1466–1471
Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G and Testi R (2000) GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 14: 2047–2054
Malisan F, Franchi L, Tomassini B, Ventura N, Condo I, Rippo MR, Rufini A, Liberati L, Nachtigall C, Kniep B and Testi R (2002) Acetylation suppresses the proapoptotic activity of GD3 ganglioside. J. Exp. Med. 196: 1535–1541
Wang HJ, Guay G, Pogan L, Sauve R and Nabi IR (2000) Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell Biol. 150: 1489–1498
Tomassini B, Malisan F, Franchi L, Nicolo C, Calvo GB, Saito T and Testi R (2004) Calnexin suppresses GD3 synthase-induced apoptosis. FASEB J. 18: 1553–1555
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219
Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T and Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20: 2690–2701
Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N and Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 97: 5723–5728
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T and Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102: 105–110
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P and Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 166: 193–203
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J and Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 162: 59–69
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J Yuan
Rights and permissions
About this article
Cite this article
d'Azzo, A., Tessitore, A. & Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ 13, 404–414 (2006). https://doi.org/10.1038/sj.cdd.4401834
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401834
Keywords
This article is cited by
-
Effect of Transmembrane Electric Field on GM1 Containing DMPC–Cholesterol Monolayer: A Computational Study
The Journal of Membrane Biology (2020)
-
Lysosomal storage diseases
Nature Reviews Disease Primers (2018)
-
An LC–MS-based lipidomics pre-processing framework underpins rapid hypothesis generation towards CHO systems biotechnology
Metabolomics (2018)
-
Detailed Structural Characterization of Sphingolipids via 193 nm Ultraviolet Photodissociation and Ultra High Resolution Tandem Mass Spectrometry
Journal of the American Society for Mass Spectrometry (2017)
-
Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms
Acta Neuropathologica (2017)
